
El síndrome de Peutz-Jeghers (SPJ) es una enfermedad de patrón hereditario autosómico dominante caracterizado por la presencia de pólipos hamartomatosos y pigmentación mucocutánea, su prevalencia es de uno por cada 8,300 a 29,000 nacidos vivos; las áreas del tracto gastrointestinal afectadas con mayor frecuencia son el intestino delgado, el yeyuno y el íleon (65-95%), y en menor grado el colon (60%) y el estómago (50%), y puede dar lugar a complicaciones como la obstrucción de la luz intestinal, el sangrado y el elevado riesgo de neoplasias intestinales y extraintestinales1-3.
Presentamos el caso de un paciente masculino de 30 años de edad, atendido en el Instituto de Investigaciones Médico-Biológicas de la Universidad Veracruzana con antecedente familiar de padre con pólipos intestinales y cuadro de un año de evolución caracterizado dolor abdominal, náuseas, meteorismo y estreñimiento acompañado de anorexia, adinamia, pérdida de peso, encontrándose como signo relevante la presencia de manchas hipercrómicas en labio superior y paladar. Se realizó colonoscopia, que mostró la presencia de múltiples lesiones poliploides sésiles de tamaño variable entre 0.3 y 2cm a lo largo de todo el colon y en el íleon terminal. El estudio histopatológico reportó pólipos hamartomatosos pediculados con componente histológico estromal y epitelial, con proliferación arborescente de músculo liso, integrado con ello el diagnóstico de SPJ.
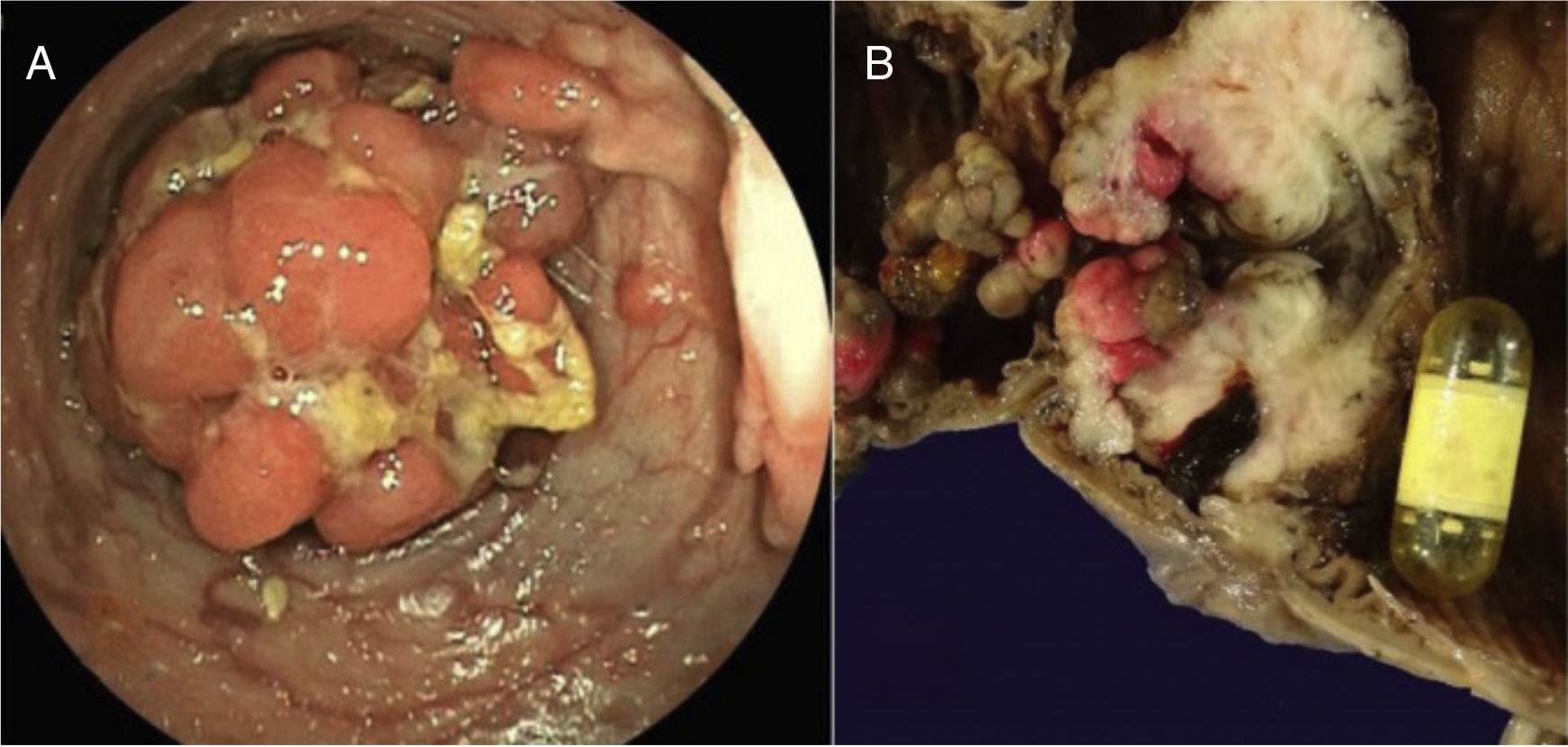
Un año después, acudió nuevamente por persistencia del dolor, ahora localizado en el flanco izquierdo, distensión abdominal, estreñimiento importante, náuseas y vómitos, y ataque al estado general; se decidió realizar estudio de cápsula endoscópica modelo Pillcam (Given Imaging), la cual mostró en la cámara gástrica, el duodeno, el yeyuno y el íleon la presencia de múltiples pólipos sésiles, sin lograr avanzar a colon debido a la presencia de un pólipo de gran tamaño. En los siguientes 7 días se intensificó el dolor, acompañado de síntomas y signos francos de obstrucción intestinal sin expulsión de la cápsula, corroborados por una placa simple de abdomen, y la presencia de la cápsula en la zona correspondiente al íleon, tomándose la decisión de realizar enteroscopia anterógrada de doble balón, logrando visualizarse múltiples pólipos ramificados, erosionados, ulcerados con sangrado en capa, encontrándose la cápsula detenida en el íleon y un pólipo de gran tamaño que no permitió el paso del endoscópico, corroborándose la obstrucción intestinal (fig. 1 A). Las maniobras para tratar de extraerla fueron infructuosas debido al impacto de la misma sobre el sitio estenosado, por lo cual el paciente fue sometido a resección quirúrgica de un segmento de 77cm de íleon terminal con ileotransversoanastomosis (fig. 1. B).
 Colonoscopia en la cual se observa la presencia de pólipo de gran tamaño. B) Pieza quirúrgica que muestra el corte de la luz del intestino con incontables pólipos y cápsula endoscópica obstruyendo su luz en una zona de estenosis en el íleon.")
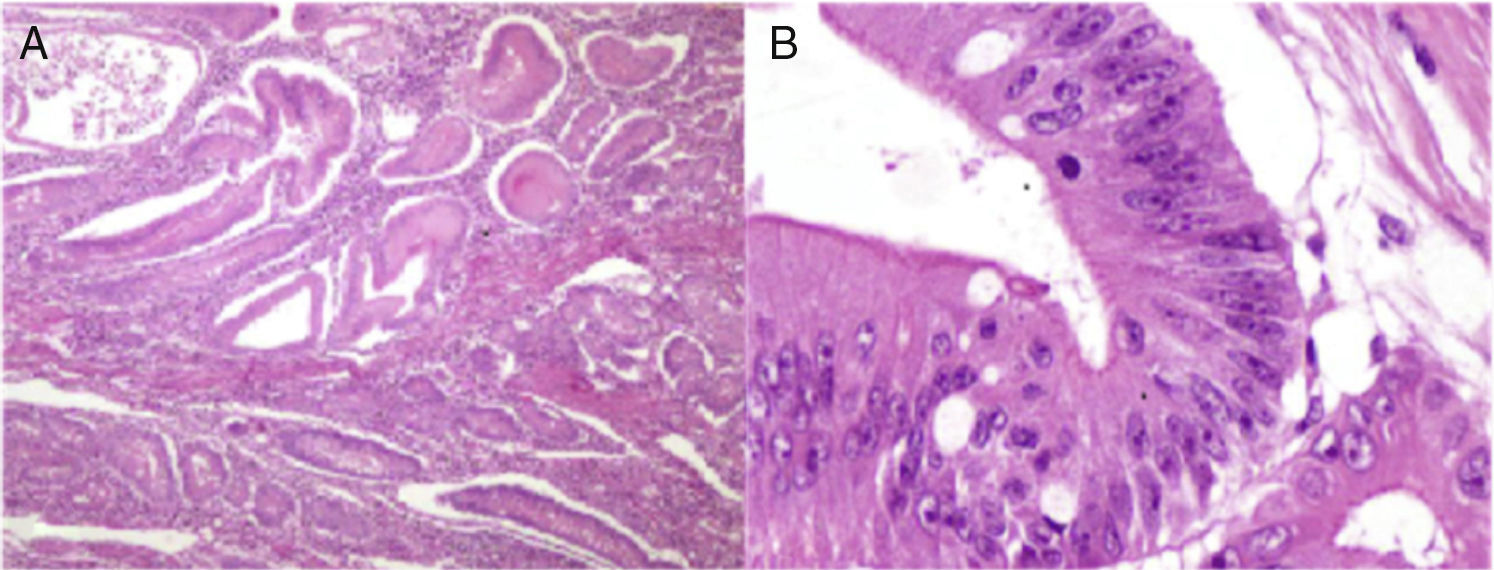
El estudio histopatológico de la pieza mostró incontables pólipos de diverso tamaño desde 0.2 hasta 5cm; el mayor de ellos obstruía el 90% de la luz, encontrándose la cápsula en la parte proximal de la lesión. El estudio microscópico reveló pólipos múltiples con hemorragia y ulcerados, y la lesión estenosada correspondió a un pólipo con displasia de alto grado focal y un aspecto seudoinvasivo estromal (fig. 2 A y B).
 Fotomicrografía a bajo aumento; se observan numerosos pólipos de Peutz-Jeghers que presentan de manera característica gruesas bandas de músculo liso entre los islotes de tejido epitelial. B) Fotomicrografía a gran aumento del pólipo de mayor tamaño que presenta displasia de alto grado")
Estudio histopatológico de la lesión. A) Fotomicrografía a bajo aumento; se observan numerosos pólipos de Peutz-Jeghers que presentan de manera característica gruesas bandas de músculo liso entre los islotes de tejido epitelial. B) Fotomicrografía a gran aumento del pólipo de mayor tamaño que presenta displasia de alto grado
Posterior a la resección intestinal, el paciente presentó evento vascular cerebral de tipo isquémico en el hemisferio derecho, asociado a foramen oval permeable diagnosticado por ultrasonido transesofágico, el cual ocasionó disminución de la fuerza y la sensibilidad en las extremidades izquierdas. La evolución posterior fue satisfactoria, con mejoría considerable de su estado general, tránsito intestinal normal y sometido a rehabilitación física, con buena respuesta neuromuscular.
Peutz describió en 1921 la relación existente entre la pigmentación mucocutánea y la poliposis intestinal al estudiar a 7 miembros de una familia. Años más tarde, en 1949, Jeghers et al. publicaron el carácter hereditario autosómico dominante y el riesgo elevado de cáncer que presentan los pacientes con dicha patología; sin embargo; no fue hasta 1954 cuando Bruwer et al. acuñaron por primera vez el término de «síndrome de Peutz-Jeghers»4. Su incidencia es baja, sin predominio de género ni raza; la edad promedio de diagnóstico es entre los 9 a 39 años, con una media de 23 años5. El 75% de los casos es resultado de la herencia dominante de la mutación genética presente en el cromosoma 19p 13.3, que codifica una serina-treonina-cinasa conocida como STK11/LKB1, y aunque su función no ha sido bien determinada, se cree que tiene un papel en desarrollo de arquitectura celular, originando una desorganización tisular de penetrancia variable que da origen a la formación de pólipos de localización variable, así como presentación de manchas hipercrómicas6,7.
Nuestro caso se trató de un paciente joven que cumplió con los criterios establecidos por la Organización Mundial de la Salud (presencia de 3 pólipos de Peutz-Jeghers confirmados por estudio histológico, con historia familiar de SPJ y con pigmentación mucocutánea)8, el cual presentó un cuadro de oclusión intestinal a nivel del íleon terminal por la enfermedad polipósica, en la cual se impactó la cápsula endoscópica, situación poco frecuente y que requirió resección intestinal del segmento afectado y que presentó un accidente vascular cerebral en el postoperatorio debido a un embolo séptico a través del foramen oval, y consideramos que no guarda relación con la patología gastrointestinal.
Se ha descrito en la literatura por varios autores que los pacientes jóvenes con SPJ tienen un elevado riesgo de desarrollar cáncer del tracto digestivo (15-18 veces mayor que la población general), por lo cual nuestro enfermo deberá ser sometido a seguimiento endoscópico con el objeto de detectar en forma temprana la posible malignización de los pólipos hamartomatosos9,10.
FinanciaciónSe utilizaron únicamente recursos institucionales.
Conflicto de interesesNo existe conflicto de intereses económicos, profesionales o personales de ninguno de los autores.

