
La enfermedad hepática alcohólica (EHA) es una entidad clinicopatológica ocasionada por el consumo excesivo y crónico de alcohol. La enfermedad incluye un amplio espectro de anomalías celulares y tisulares que pueden causar daño agudo sobre crónico (hepatitis alcohólica) o crónico (fibrosis, cirrosis, cáncer hepatocelular), teniendo un gran impacto en la morbimortalidad en todo el mundo. El alcohol es metabolizado principalmente en el hígado. Durante el metabolismo del alcohol son generados metabolitos tóxicos como el acetaldehído y las especies reactivas de oxígeno. En el intestino el consumo de alcohol puede producir disbiosis y alteración de la permeabilidad intestinal, promoviendo la translocación de productos bacterianos y provocando la producción de citocinas proinflamatorias en el hígado, lo cual perpetúa la inflamación local durante la evolución de la EHA. Diferentes grupos de estudio han reportado alteraciones de la respuesta inflamatoria a nivel sistémico, sin embargo es difícil encontrar reportes que contengan un compendio de las células y citocinas involucradas en la fisiopatología de la enfermedad desde sus etapas tempranas. En este trabajo de revisión se describe el papel de los mediadores inflamatorios involucrados en la progresión de la EHA, desde los patrones de consumo de riesgo hasta etapas avanzadas de la enfermedad, con la finalidad de comprender la implicación de la desregulación inmunológica en la fisiopatología de esta enfermedad.
Alcoholic liver disease (ALD) is a clinical-pathologic entity caused by the chronic excessive consumption of alcohol. The disease includes a broad spectrum of anomalies at the cellular and tissual level that can cause acute-on-chronic (alcoholic hepatitis) or chronic (fibrosis, cirrhosis, hepatocellular cancer) injury, having a great impact on morbidity and mortality worldwide. Alcohol is metabolized mainly in the liver. During alcohol metabolism, toxic metabolites, such as acetaldehyde and oxygen reactive species, are produced. At the intestinal level, alcohol consumption can cause dysbiosis and alter intestinal permeability, promoting the translocation of bacterial products and causing the production of inflammatory cytokines in the liver, perpetuating local inflammation during the progression of ALD. Different study groups have reported systemic inflammatory response disturbances, but reports containing a compendium of the cytokines and cells involved in the pathophysiology of the disease, from the early stages, are difficult to find. In the present review article, the role of the inflammatory mediators involved in ALD progression are described, from risky patterns of alcohol consumption to advanced stages of the disease, with the aim of understanding the involvement of immune dysregulation in the pathophysiology of ALD.
La enfermedad hepática alcohólica (EHA) es una entidad clínico-patológica ocasionada por el consumo excesivo de alcohol, la cual puede incluir un espectro de entidades tales como esteatosis, esteatohepatitis, fibrosis, cirrosis, carcinoma hepatocelular y, en algunos casos, hepatitis alcohólica (HA). La progresión de una entidad a otra depende de la continuación del consumo de alcohol y de la interacción compleja de un gran número de variables: genéticas, biológicas, inmunológicas, psicológicas, socioculturales y ambientales; sin embargo, aún no se ha esclarecido por completo su patogenia1,2.
En México, los resultados de la Encuesta nacional de consumo de drogas, alcohol y tabaco (ENCODAT, 2016-2017), revelan que la población mexicana entre 18 a 65 años ha tenido un incremento en el consumo de alcohol, observándose de manera marcada un aumento en la dependencia a dicha sustancia en mujeres adolescentes3. La edad de inicio de consumo de bebidas alcohólicas pasó de 17 a 16 años, reflejando que los adolescentes son un grupo de riesgo para desarrollar trastornos relacionados con el abuso del alcohol3.
El consumo de alcohol en estos grupos etarios constituye un factor de riesgo para el desarrollo de adicción, ya que se induce alteración a nivel cerebral en la región perifrontal, la cual es la encargada de la planificación y regulación conductual4,5. Respecto a la influencia genética se ha descrito que ADH1B y ALDH2 son los 2 genes principales que participan en el metabolismo del alcohol, los cuales están relacionados con el consumo excesivo y con la dependencia a dicha sustancia; la información de estos genes y sus variantes genéticas está descrita de manera excelente en revisiones especializadas del tema6,7, sin embargo aún no se utilizan como herramienta para el diagnóstico del consumo de alcohol en sus diferentes patrones.
En México la EHA es la causa más frecuente de cirrosis hepática8. Mientras que la tasa de incidencia de cirrosis hepática alcohólica hasta junio del año 2021 fue de 2 casos por cada 100,000 habitantes, situando a México en el quinto lugar de mortalidad por enfermedad hepática9, y de acuerdo con estudios de proyecciones de la enfermedad, se espera que la mortalidad aumente en los siguientes años10.
El grupo etario más afectado de cirrosis por EHA es el de 51 a 70 años, debido a que generalmente se busca atención médica hasta etapas avanzadas de la enfermedad11. Debido al consumo activo de alcohol en la población mexicana y a la baja tasa de tamizaje para el consumo riesgoso de alcohol y, en consecuencia, el desarrollo de la EHA, se prevé que el número de casos de daño hepático y cirrosis aumente en los siguientes años, incrementando la mortalidad de dicha enfermedad8. Extensas descripciones epidemiológicas en México y en el mundo han sido descritas de manera detallada por varios autores12–14. En el mundo la mayoría de los casos de EHA se diagnostican en estadios avanzados de la enfermedad, es decir, cirrosis hepática11,15. Hasta la fecha para valorar el consumo de alcohol se utilizan cuestionarios que permiten realizar una evaluación psiquiátrica16, sin embargo no se cuenta con una herramienta cuantitativa y objetiva.
La EHA puede cursar con etapas agudas y crónicas, o incluso con una combinación de ambas; es por ello que la respuesta inmunológica, local (hígado) y sistémica (circulación sanguínea) participa de manera activa de acuerdo con la frecuencia y el tipo de alcohol consumido1,17. En este trabajo de revisión se hace énfasis en los efectos fisiopatológicos en la EHA y las alteraciones inmunológicas durante la progresión de la enfermedad. Además, se exponen los criterios de clasificación del consumo de alcohol, los cuales no han sido del todo definidos, y son de suma importancia para el diagnóstico temprano de enfermedad por abuso en el consumo de alcohol.
Patrón de consumo de alcoholLa bebida estándar hasta ahora tiene mundialmente varias definiciones, por lo que la cantidad y frecuencia de consumo suele ser un aspecto de difícil determinación. En México la bebida estándar tiene 13g de alcohol18. En cuanto al tipo de bebida alcohólica consumida los resultados de la ENCODAT reportaron que los fermentados, específicamente la cerveza, ocupan el primer lugar de prevalencia con un 40.8%, y en segundo lugar se encuentran los destilados (brandy, tequila, ron, whisky, coñac, vodka, etc.) con un 19.1%3. La calidad de las bebidas alcohólicas aún es tema de controversia, ya que no tiene una regulación en cuanto a sus estándares; finalmente, los efectos a nivel clínico y el daño hepático suelen ser evidentes hasta etapas avanzadas de la EHA.
El consumo moderado de alcohol se ha asociado con efectos protectores cardiovasculares, efectos antidepresivos y reducción de accidentes cerebrovasculares19–21, siendo definido el consumo moderado como la ingesta de una bebida estándar al día para las mujeres y 2 para los hombres22. Se considera que un individuo tiene un consumo riesgoso de alcohol cuando tiene un puntaje de 8 o superior en el cuestionario de identificación de trastornos por consumo de alcohol (AUDIT)23, e ingiere entre 40-60g (en los hombres) o 20-40g (en las mujeres)24. El abuso en el consumo de alcohol es usualmente considerado cuando el cuestionario AUDIT tiene un puntaje de 8 o superior, y se encuentra en los criterios de abuso según el Manual diagnóstico y estadístico de los trastornos mentales (DSM)-IV25. Este patrón se caracteriza por una ingesta superior a 60g (en los hombres) o mayor a 40g (en las mujeres), además el consumo consuetudinario es>70g en hombres y>50g en mujeres, de una hasta 4 veces por semana.
El alcoholismo, de acuerdo con la Organización Mundial de la Salud (OMS), se define como la ingesta diaria de más de 70g (en los hombres) y de 50g (en las mujeres), por al menos 5 años, con un puntaje del AUDIT mayor a 8, y que cumple con criterios de dependencia según el DSM-IV26–28. Actualmente es común encontrar sujetos diagnosticados como alcohólicos que beben esas cantidades en fin de semana o de manera consuetudinaria29. Es importante mencionar que los individuos con patrones de consumo riesgoso, abuso y alcoholismo no reportan alteraciones evidentes en los parámetros clínicos y bioquímicos, y por tal motivo es difícil que estos sujetos acudan al médico11.
Dentro de las herramientas actuales para el diagnóstico de problemas con el consumo de alcohol se encuentra el AUDIT, CAGE (Cut down, Annoyed, Guilty and Eye-opener questions) y el DSM; sin embargo, hasta la fecha no existen parámetros bioquímicos o marcadores específicos que permitan determinar cuantitativamente el efecto del consumo nocivo de alcohol desde una etapa temprana, como lo es la esteatosis hepática. Frecuentemente los pacientes son asintomáticos hasta que desarrollan una etapa grave y avanzada1.
Nuestro grupo de trabajo ha encontrado que los individuos que ya han desarrollado EHA (cirrosis o HA) consumen con mayor frecuencia destilados, alcohol de caña y/o alcohol de 96° (trabajo en proceso). El índice Maddrey es el sistema de puntuación usado más ampliamente en HA; está basado en los valores de bilirrubina y el tiempo de protrombina al ingreso del paciente30,31. Este funciona como una guía para el tratamiento y como un predictor de la mortalidad y la severidad de la enfermedad30,31. Un puntaje>32 indica que la enfermedad es grave, y conlleva una alta mortalidad (30-60%) a corto plazo, mientras que<32 indica una enfermedad no grave con una mortalidad a corto plazo del 10%30,31. Por otro lado, la cirrosis hepática por alcohol es una condición crónica que presenta signos característicos como sarcopenia, pérdida de peso, astenia, adinamia, reflujo hepatoyugular, telangiectasias, ginecomastia y eritema palmar, entre otros1. Sin embargo, algunos pacientes con cirrosis son diagnosticados cuando desarrollan descompensación hepática, frecuentemente en el contexto de una HA superpuesta1. La escala de Child-Pugh ha sido utilizada ampliamente para evaluar la supervivencia de pacientes con cirrosis32. Se clasifican en clase A si la puntuación es de 5-6 puntos, clase B si es de 7-9 puntos y clase C si es de 10-15 puntos, donde la clase A tiene la mayor supervivencia al cabo de un año (100%) y 2 años (85%) y la case C tiene la peor supervivencia al año (45%) y a los 2 años (35%)32.
Fisiopatología en la enfermedad hepática alcohólicaEl alcohol es metabolizado de manera primaria en el estómago y en el intestino, en donde se forman metabolitos tóxicos como el acetaldehído, un componente fundamental para la progresión de la EHA33,34. Posteriormente, el alcohol pasa al torrente sanguíneo y a la circulación portal para llegar al hígado, donde se llevará a cabo la mayor parte del metabolismo mediante los hepatocitos; si el consumo es excesivo se activa la vía microsómica, lo que puede generar especies reactivas de oxígeno (ERO)33,35,36.
Durante la EHA la esteatosis se presenta en alrededor del 90% al 100% de los consumidores de alcohol, y el 35% de ellos progresan a esteatohepatitis; cuando el consumo excesivo de alcohol continúa se desarrolla fibrosis hepática, la cual puede avanzar a cirrosis en alrededor del 20% de los casos; del 1% al 2% de estos últimos desarrollará hepatocarcinoma17. No obstante, no se trata de etapas separadas de la enfermedad, sino que pueden coexistir en la misma persona17,37. Por su parte, la HA se puede desarrollar en el 40% de los sujetos con cirrosis; no obstante, esta puede aparecer, incluso, desde esteatohepatitis por un consumo excesivo de alcohol2,17. La HA es una complicación grave que puede ocurrir en cualquier punto en el curso de la enfermedad, y se asocia con insuficiencia hepática y con una mortalidad a corto plazo de hasta el 40%38,39. A pesar de que existen etapas bien definidas, la interacción compleja entre los factores celulares y moleculares de la EHA aún no es del todo comprendida2,17.
Eje intestino-hígadoEl consumo excesivo de alcohol puede provocar digestión deficiente, mala absorción de nutrientes, deficiencias de vitaminas y oligoelementos y, por lo tanto, pérdida de peso, debido principalmente al metabolismo de primer paso del alcohol40. La toxicidad del alcohol está dada principalmente por el producto metabólico del alcohol, el acetaldehído, que además puede ser producido por varias bacterias del colon, lo que conduce al daño de las células de la mucosa colónica41.
El acetaldehído al nivel del colón puede provocar alteración de las proteínas de las uniones estrechas, promoviendo disfunción de la barrera intestinal41. Además, el consumo de alcohol puede afectar la motilidad intestinal, el pH de la luz intestinal y el flujo de bilis42.
EsteatosisEn el hígado la alteración inicial en la EHA es la esteatosis, debido a una desregulación del recambio de lípidos17,43. El alcohol puede inducir esteatosis mediante las siguientes alteraciones:
- -
Elevación de la proporción de NADH/NAD+ en los hepatocitos, interrumpiendo la β-oxidación de los ácidos grasos en las mitocondrias44.
- -
Aumento de la expresión hepática de SREBP1c (factor de unión a elementos de respuesta a esteroles), estimulando la expresión de genes lipogénicos y la síntesis de ácidos grasos45,46. La alteración de la transcripción de SREBF1 que codifica SREBP1c y PPARα que codifica PPARα (receptor alfa activado por proliferador de peroxisomas), puede ocurrir directamente por el acetaldehído, aunque también se ha encontrado que puede activarse por la translocación bacteriana47.
- -
Inhibición de la proteína cinasa activada por AMP, promoviendo la síntesis de ácidos grasos e inhibiendo la β-oxidación mediante la desregulación de la acetil-CoA carboxilasa, la isoforma hepática de la carnitina palmitoiltransferasa 1 y SREBP48.
- -
Inducción de lipólisis y destrucción de adipocitos, lo que resulta en la elevación de los ácidos grasos circulantes y su posterior acumulación en el hígado43,49,50.
Otra etapa bien definida es la esteatohepatitis alcohólica, caracterizada por la presencia de lípidos y una evidente reacción inflamatoria, con predominio de neutrófilos, degeneración balonizante de los hepatocitos, cuerpos de inclusión de Mallory-Denk y una red de fibrosis perivenular51. Cabe destacar que las reacciones inflamatorias se inducen desde la esteatosis, sin embargo se incrementan de manera franca durante la esteatohepatitis alcohólica52. La actividad inmunitaria produce estrés del retículo endoplasmático, activando el factor regulador de interferón 3 (IRF3); no obstante, la molécula IRF3 se fosforila ante la exposición al alcohol, lo que activa la apoptosis de los hepatocitos41. Las células de Kupffer son activadas por la traslocación del lipopolisacárido (LPS), promoviendo el reclutamiento de neutrófilos, debido a que las células de Kupffer secretan citocinas proinflamatorias tales como TNF-α y la quimiocina CXCL-853.
Factores que contribuyen al desarrollo de fibrosis y cirrosisLa fibrosis es la etapa caracterizada por una acumulación descontrolada de matriz extracelular en el ámbito pericelular y perisinusoidal como respuesta al daño e inflamación persistente1. La gran cantidad de citocinas y factores de crecimiento secretados por las células de Kupffer promueven la activación de las células estelares hepáticas (CEH), siendo este el evento clave en la fibrogénesis hepática1. El acetaldehído por sí mismo es capaz de inducir directamente la activación de las CEH54,55. El consumo excesivo de alcohol interviene en la regulación de las CEH, al suprimir la actividad citotóxica de las células natural killer (NK) hacia las CEH activadas, además de su secreción de IFN-γ que induce la apoptosis de las CEH activadas; favoreciendo la producción excesiva de matriz extracelular por las CEH56.
No obstante, la fibrosis del hígado es multifactorial, y entre los factores involucrados en su desarrollo se encuentran las ERO provenientes del metabolismo del alcohol que producen peroxidación lipídica, obteniendo como resultado metabolitos como malondialdehído y 4-hidroxi-nonenal, los cuales inducen la formación de neoantígenos, alterando la respuesta inmune57. El acetaldehído ingresa en las CEH de manera paracrina e induce la expresión de TGF-β1, y en consecuencia se estimula la producción de colágeno tipo i57. El acetaldehído fosforila la proteína SMAD 3, lo que produce la activación del complejo SMAD 3-4; es así como el TGF-β y el acetaldehído forman una retroalimentación positiva de los genes de colágeno, en especial del gen α2 de colágeno tipo i58.
Por otra parte, se ha observado que la apoptosis de los hepatocitos tiene un papel en la aparición de fibrosis. Los cuerpos apoptóticos liberan señales lipídicas para su procesamiento por las células de Kupffer y las CEH, promoviendo la expresión de genes profibrogénicos, como TGF-β1, que desencadenan la activación de las CEH58. Los mecanismos antes descritos pueden disminuir si el paciente deja de consumir bebidas alcohólicas, pero si el proceso inflamatorio continúa se progresa a una etapa crónica con una fibrogénesis sostenida, lo que resulta en la sustitución del parénquima hepático por tejido cicatricial que compromete la arquitectura y la red vascular del hígado, desarrollando cirrosis que se caracteriza por la formación de nódulos regenerativos del parénquima hepático rodeados de tabiques fibrosos59,60. Su desarrollo consiste en una fase compensada, donde una parte del hígado permanece intacta y compensa funcionalmente las regiones dañadas; posteriormente, avanza a una fase descompensada, en la que el tejido cicatricial envuelve completamente el órgano61,62. Este último se caracteriza por el desarrollo de hipertensión portal con o sin presencia de insuficiencia hepática63.
Uso de antiinflamatorios en la enfermedad hepática alcohólicaHasta la fecha no existe un tratamiento que involucre moléculas antiinflamatorias para la hepatopatía por alcohol64. La entidad más estudiada es la HA debido a su alta mortalidad en poco tiempo (28 días), y desde hace más de 50 años los corticosteroides han tenido un papel central; sin embargo, su beneficio en el tratamiento se ha vuelto controvertido65. El ensayo clínico aleatorizado más grande en HA Steroids or Pentoxifylline for Alcoholic Hepatitis (STOPAH), no demostró un beneficio estadísticamente significativo en la supervivencia a los 28 días en pacientes que recibieron corticosteroides en comparación con placebo (p=0.06). Por su parte, las guías vigentes de manejo de HA recomiendan la administración oral de 40mg al día de prednisona para reducir la mortalidad a los 28 días en pacientes con HA grave66. Por lo antes mencionado, existe una necesidad ineludible de una mejoría en el tratamiento de HA y EHA. En este sentido, el IMM-124E es una formulación de calostro bovino hiperinmune purificado, la cual contiene IgG contra LPS, y fue desarrollado con la finalidad de reducir la endotoxemia; los resultados del ensayo clínico aleatorizado (NCT01968382) no mostraron que tuviera eficacia como terapia adyuvante a los corticoides en pacientes con HA grave65. Por su parte, anakinra es un antagonista del receptor de IL-1; en ensayos clínicos (NCT01809132) de fase ii y iii se comparó la combinación de anakinra, pentoxifilina y sulfato de cinc frente al tratamiento con metilprednisolona67, no obstante no se observó una mejoría en la supervivencia en la combinación terapéutica evaluada67,68.
El uso de antagonistas de TNF-α como infliximab y etanercept se ha asociado a un aumento de la mortalidad y de la incidencia de infecciones69,70, por lo que las guías vigentes ya no lo recomiendan66. Recientemente, el ensayo clínico de fase ii, utilizando F652 (proteína recombinante), mostró una disminución en el puntaje MELD, así como de los marcadores de inflamación, y un incremento en los marcadores de regeneración hepática en pacientes con HA71.
Por otra parte, dentro de las terapias que tienen un beneficio potencial con eficacia clínica, pero que no han sido probadas del todo, se encuentran las terapias con antioxidantes tales como la N-acetilcisteína (NAC) y la metadoxina65,66,72–74. En un ensayo clínico aleatorizado en pacientes con HA grave, la infusión de NAC como una terapia adyuvante a los corticosteroides mejoró la sobrevida al mes, sin embargo no se observó un beneficio a largo plazo, esto último debido principalmente al aumento en las tasas de infección y al desarrollo del síndrome hepatorrenal73. Por otro lado, la metadoxina como una terapia adyuvante a los corticosteroides o a la pentoxifilina se asoció a una mejoría en la supervivencia a los 3 y 6 meses en pacientes con HA grave74.
Finalmente, el trasplante hepático es el procedimiento indicado en daño hepático avanzado, sin embargo un alto porcentaje de pacientes fallece antes de entrar a la lista de espera10,75,76.
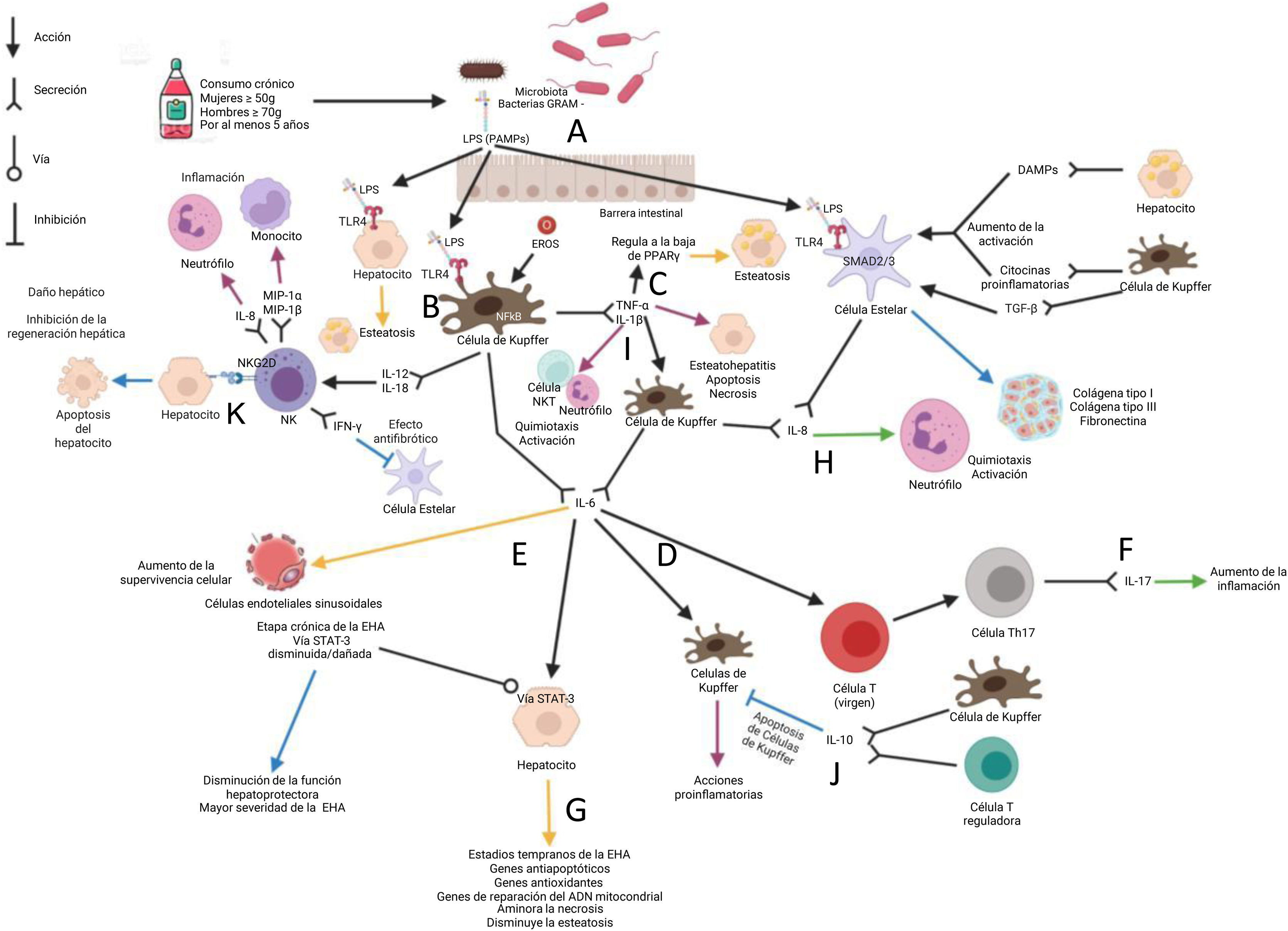
Inflamación durante la progresión de la enfermedad hepática alcohólicaEl consumo crónico de alcohol da lugar a disbiosis intestinal, provocando un cambio de dominancia hacia bacterias principalmente Gram (–) y agentes oportunistas; así mismo, causa una disminución de los microorganismos que caracterizan la mucosa intestinal de sujetos sanos40. La mucosa duodenal de sujetos con un consumo crónico de alcohol presenta una disminución de células inflamatorias como macrófagos y linfocitos T, por lo tanto presentan una respuesta inflamatoria atenuada, siendo este último un posible factor permisivo de la translocación bacteriana40. Así mismo, existe una disrupción de las proteínas de las uniones estrechas presentes en la barrera intestinal, dando lugar a una permeabilidad intestinal incrementada40 (fig. 1A).
 (A). La ingesta crónica de alcohol da lugar a la disrupción de la barrea intestinal, provocando el aumento de la permeabilidad intestinal, lo que permite la translocación de LPS y su llegada al hígado a través de la circulación portal. En el hígado el LPS se une al TLR4 en las células de Kupffer (B) que, en conjunto con ERO, estimulan la síntesis de TNF-α e IL-1β. TNF-α regula a la baja la expresión del gen PPARɣ, favoreciendo la expresión de genes que participan en la β-oxidación, contribuyendo al desarrollo de esteatosis (C). TNF-α e IL-1β dan lugar a la producción de IL-6 por las células de Kupffer y, por lo tanto, perpetúan el estado inflamatorio (D). La producción de IL-6 por parte de las células de Kupffer desempeña un papel dual, favoreciendo la inflamación a través de la retroalimentación positiva de las células de Kupffer (E) y la activación de la respuesta Th17 (F). Por otra parte, IL-6 favorece la activación de STAT-3 en etapas tempranas de la EHA, dando lugar a un efecto hepatoprotector (G). La IL-8 provoca la quimiotaxis y activación de neutrófilos, provocando inflamación y daño tisular (H). La estimulación de las células estelares por PAMPs, DAMPs y las células de Kupffer favorece su activación y posteriormente la producción de IL-8 y componentes de la matriz extracelular, dando lugar al desarrollo de fibrosis (I). La IL-10 regula negativamente las respuestas inflamatorias a través de promover la apoptosis de macrófagos con perfil inflamatorio (J). Las células de Kupffer activadas a través de la unión de ligandos a su receptor TLR4 inducen el reclutamiento y la activación de las células NK, mediante la producción de IL-12 e IL-18; siendo estas últimas citocinas fuertes inductoras de la producción de IFN-y por las células NK, el cual tiene un efecto antifibrótico al inducir la apoptosis y el arresto del ciclo celular de las CEH, así como al incrementar la citotoxicidad de las NK (K). Flecha amarilla: esteatosis; flecha morada: esteatohepatitis; flecha azul: fibrosis; flecha verde: hepatitis alcohólica. ADN: ácido desoxirribonucleico (imagen diseñada con BioRender.com); DAMPs: patrones moleculares asociados a daño; EHA: enfermedad hepática alcohólica; ERO: especies reactivas de oxígeno; IFN-γ: interferón gamma; IL: interleucina; LPS: lipopolisacárido; MIP-1α: proteína inflamatoria macrofágica 1 alfa; MIP-1β: proteína inflamatoria macrofágica 1 beta; NF-kB: factor nuclear kappa B; NK: células natural killer; NKT: células natural killer T; NKG2D: natural killer group 2D receptor; PAMPs: patrones moleculares asociados a patógenos; TGF- β: factor de crecimiento transformante beta; TLR4: receptor tipo-Toll 4; TNF-α: factor de necrosis tumoral alfa; PPARα: receptor activado por proliferadores peroxisómicos tipo alfa; SMAD: familia Smad; STAT3: transductor de señal y activador de la transcripción 3.")
El consumo crónico de alcohol ocasiona disbiosis intestinal, favoreciendo la proliferación de bacterias Gram (–) (A). La ingesta crónica de alcohol da lugar a la disrupción de la barrea intestinal, provocando el aumento de la permeabilidad intestinal, lo que permite la translocación de LPS y su llegada al hígado a través de la circulación portal. En el hígado el LPS se une al TLR4 en las células de Kupffer (B) que, en conjunto con ERO, estimulan la síntesis de TNF-α e IL-1β. TNF-α regula a la baja la expresión del gen PPARɣ, favoreciendo la expresión de genes que participan en la β-oxidación, contribuyendo al desarrollo de esteatosis (C). TNF-α e IL-1β dan lugar a la producción de IL-6 por las células de Kupffer y, por lo tanto, perpetúan el estado inflamatorio (D). La producción de IL-6 por parte de las células de Kupffer desempeña un papel dual, favoreciendo la inflamación a través de la retroalimentación positiva de las células de Kupffer (E) y la activación de la respuesta Th17 (F). Por otra parte, IL-6 favorece la activación de STAT-3 en etapas tempranas de la EHA, dando lugar a un efecto hepatoprotector (G). La IL-8 provoca la quimiotaxis y activación de neutrófilos, provocando inflamación y daño tisular (H). La estimulación de las células estelares por PAMPs, DAMPs y las células de Kupffer favorece su activación y posteriormente la producción de IL-8 y componentes de la matriz extracelular, dando lugar al desarrollo de fibrosis (I). La IL-10 regula negativamente las respuestas inflamatorias a través de promover la apoptosis de macrófagos con perfil inflamatorio (J). Las células de Kupffer activadas a través de la unión de ligandos a su receptor TLR4 inducen el reclutamiento y la activación de las células NK, mediante la producción de IL-12 e IL-18; siendo estas últimas citocinas fuertes inductoras de la producción de IFN-y por las células NK, el cual tiene un efecto antifibrótico al inducir la apoptosis y el arresto del ciclo celular de las CEH, así como al incrementar la citotoxicidad de las NK (K).
Flecha amarilla: esteatosis; flecha morada: esteatohepatitis; flecha azul: fibrosis; flecha verde: hepatitis alcohólica.
ADN: ácido desoxirribonucleico (imagen diseñada con BioRender.com); DAMPs: patrones moleculares asociados a daño; EHA: enfermedad hepática alcohólica; ERO: especies reactivas de oxígeno; IFN-γ: interferón gamma; IL: interleucina; LPS: lipopolisacárido; MIP-1α: proteína inflamatoria macrofágica 1 alfa; MIP-1β: proteína inflamatoria macrofágica 1 beta; NF-kB: factor nuclear kappa B; NK: células natural killer; NKT: células natural killer T; NKG2D: natural killer group 2D receptor; PAMPs: patrones moleculares asociados a patógenos; TGF- β: factor de crecimiento transformante beta; TLR4: receptor tipo-Toll 4; TNF-α: factor de necrosis tumoral alfa; PPARα: receptor activado por proliferadores peroxisómicos tipo alfa; SMAD: familia Smad; STAT3: transductor de señal y activador de la transcripción 3.
El LPS derivado de bacterias llega al hígado a través de la circulación portal, interactúa con el TLR4 en las células de Kupffer, activando la cascada de señalización de NF-kB, y dando lugar a la producción de citocinas proinflamatorias como TNF-α e IL-1β41 (fig. 1B). En el consumo crónico de alcohol el metabolismo del etanol se encuentra aumentado, produciendo acetaldehído y ERO que inducen la activación de las células de Kupffer, las cuales desencadenan la producción de un ambiente proinflamatorio (TNF-α, IL-1, IL-6, IL-8, MCP-1 y TGF-β) y de ERO como el anión superóxido, metabolitos del ácido araquidónico (prostaglandina D2, tromboxano A2, prostaglandina E2 y leucotrienos), favoreciendo el daño y/o la muerte hepatocelular36.
Se ha descrito una correlación positiva de los niveles séricos de TNF-α con el sistema de puntuación Child-Pugh en pacientes con cirrosis por alcohol77. En un modelo murino de ratones Sprague-Dawley, a los que se les administró 1μg de LPS de Escherichia coli por kilogramo de peso corporal vía intraperitoneal, se identificó que el TNF-α regula a la baja la expresión del gen PPARɣ, favoreciendo la expresión de genes que participan en la β-oxidación, contribuyendo a la esteatosis de la EHA78 (fig. 1C).
Otra citocina que ha sido descrita en el contexto de la EHA es la IL-6, la cual es una citocina pleiotrópica que exhibe funciones contrastantes: actúa como citocina proinflamatoria en modelos de enfermedades inflamatorias crónicas y, por el contrario, muestra efectos antiinflamatorios en la inflamación aguda, incluso se ha propuesto que participa en los fenómenos de regeneración hepática79,80 (figs. 1D y E). Se sugiere que la IL-6 tiene un efecto protector en la fase temprana de la EHA al participar en la supresión de quimiocinas que atraen principalmente neutrófilos y monocitos81–83. Además, puede promover la diferenciación de Th17 y la producción de IL-17, contribuyendo a la inflamación del hígado inducida por etanol84 (fig. 1F). Los niveles aumentados de IL-6 se han correlacionado con la gravedad y la cronicidad de la EHA85. La IL-6 puede proteger contra la apoptosis de los hepatocitos y participar en la reparación del ADN mitocondrial después de una lesión hepática alcohólica86,87 (fig. 1G). Es importante determinar las etapas, concentraciones y el microambiente molecular que desencadenan que la IL-6 pueda tener uno u otro papel durante la EHA.
Por otra parte, en cultivos primarios de hepatocitos de ratas alimentadas de manera crónica con etanol se encontró aumentada la citotoxicidad por TNF-α, así como la susceptibilidad al daño mitocondrial desencadenando procesos de apoptosis y necrosis88. Así mismo, se ha identificado en estos hepatocitos primarios que el TNF-α producido por las células de Kupffer induce la transcripción del ARNm de IL-889 (fig. 1H).
La IL-8 o CXCL-8 es una de las quimiocinas que, cada vez más, ha sido aceptada como un indicador del daño hepático en la EHA; esta quimiocina está involucrada en la movilización de neutrófilos desde la médula ósea hasta la infiltración y activación tisular90 (fig. 1H). La IL-8/CXCL-8 es producida por células de Kupffer en respuesta a TNF-α y ligandos para TLR mediante la activación de NF-kB91. Las CEH activadas secretan IL-8/CXCL-8 en respuesta a la estimulación por citocinas inflamatorias, como las producidas por las células de Kupffer y algunos linfocitos91 (fig. 1H). En pacientes con HA la expresión de estas quimiocinas en el hígado se correlaciona de manera positiva con la gravedad de la hipertensión portal y la supervivencia del paciente90,92.
Tanto en modelos animales como en pacientes con EHA los niveles de pro-IL-1β aumentan significativamente en el hígado y el suero en comparación con los controles93,94. En la EHA IL-1β participa en el reclutamiento y activación de células NKT y neutrófilos, provocando inflamación y lesión hepática95. Esta citocina induce la expresión de TGF-β por las células de Kupffer, promoviendo la activación de las CEH, las cuales comienzan la producción de componentes de la matriz extracelular, que de manera desregulada dan lugar a la progresión de eventos fibróticos94 (fig. 1I).
La IL-4 es un mediador profibrótico casi 2 veces más eficaz que el TGF-β para mediar la fibrosis96. Posee efectos inhibitorios sobre el IFN-γ77. La IL-13 se considera esencial para el desarrollo de la fibrosis, ya que esta citocina estimula la síntesis de colágeno por los fibroblastos y promueve la producción de TGF-β97. Ambas citocinas (IL-4 e IL-13) tienen el mismo receptor en los fibroblastos, los cuales producen proteínas de matriz extracelular como el colágeno tipo i, iii y fibronectina, al ser estimulados mediante dichas interleucinas96,98,99.
La IL-10 es una citocina que regula negativamente las respuestas inflamatorias a través de muchos mecanismos100. Se ha descrito un efecto antiinflamatorio en pacientes con fibrosis al promover la apoptosis de macrófagos con perfil inflamatorio101 (fig. 1J). Además, suprime la síntesis de proteínas de la matriz extracelular por los fibroblastos, indicando una posible inhibición de la fibrosis45. La descripción de las células productoras, las células blanco y los efectos celulares, así como los modelos de estudios de las citocinas y otros mediadores celulares se resumen en la tabla 1.
Citocinas que participan en la enfermedad hepática alcohólica
| Citocina | Célula productora | Célulablanco | Receptor | Función y evidencia experimental y/o clínica | Etapa de la EHA en la que participa | Modelo de estudio | Ref. |
|---|---|---|---|---|---|---|---|
| TNF-α | Células de KupfferHepatocitosCélulas endoteliales sinusoidalesCEH | Células de Kupffer | TNFR1 | Inducción del ARNm de IL-8Inducción de IL-6 e IL-1β vía NF-kB. | Inflamación | Tejido hepáticoH | Thornton et al.89 |
| Hepatocitos | Apoptosis a través de p38/ MAPK, activando la caspasa-3, la despolarización y promoviendo muerte celular programada | Esteatohepatitis | HepatocitosRa | Pastorino et al.88 | |||
| CEH | Efecto antiapoptótico y antiproliferativo en las CEH activadas. | Fibrosis | CEHR | Saile et al.102 | |||
| Promueve transdiferenciación de CEH a miofibroblastos fibrogénicos vía NF-kB | Fibrosis (F1)* | Tejido hepáticoR | Pradere et al.59 | ||||
| CEH | Producción de MMP-9, por reducción del ARNm de TIMP1Remodelación de matriz extracelular | Fibrosis (F1)* | CEHR0,0H | Tarrats et al.103 | |||
| IL-1β | Células de Kupffer | Células de Kupffer | IL-1R1 | Promueve esteatosis y daño hepáticoPromueve la expresión de los componentes del inflamosoma pro-casp-1 (Asc y Nlrp3)Reclutamiento y activación de células de Kupffer y linfocitos, favoreciendo la producción de TNF-α, IL-6, MCP-1/CCL-2 e IL-10 | Esteatosis | Tejido hepáticoR | Petrasek et al.94 |
| CEH | Activación deficiente de IL-1β vía Casp-1, resulta en reducción de fibrosis hepática y disminución de la expresión de TGF-β1 y pro-collal (fenotipo de fibrogénesis). | Fibrosis (F1)* | Tejido hepáticoR | Petrasek et al.94 | |||
| Promueve el incremento del propéptido N terminal del procolágeno tipo iii, TIMP-1 y ácido hialurónico | SueroR | ||||||
| IL-6 | Células de KupfferCélulas estromalesCélulas dendríticasFibroblastosCélulas endoteliales sinusoidalesCélulas mesenquimales | Células de Kupffer | IL-6Ra | La deficiencia de IL-6 resulta en el desarrollo de esteatosis y niveles altos de malondialdehídoAl recibir dosis exógena de IL-6 se observa reversión del fenotipo de esteatosis | Esteatosis | Tejido hepáticoR | El-Assal et al.104 |
| Células de KupfferCEHCélulas sinusoidalesCélulas endotelialesHepatocitos | sIL-6Ra acoplado congp-130IL-6R | Promueve inflamación:- Señalización trans (IL-6Ra soluble)-Proliferación y supervivencia de los linfocitos T por medio de IL-2 vía STAT3/Bcl-2- Promueve la citotoxicidad de linfocitos T CD8- Diferenciación a linfocitos Th1 y Th17Papel hepatoprotector (actividad regenerativa y/o antiinflamatoria)- Señalización clásica (IL-6Ra)- Activación de STAT3 y JAK- Refuerza la expresión del angiotensinógeno- Proliferación vía MAPK y PI3K/Akt- Transición de los hepatocitos quiescentes de fase G0 a G1 y S (regeneración)- Reduce la apoptosis de los hepatocitos por disminución de Bax y aumento de Bcl-1- Activación del sistema de reparación del ADN por inhibición del ciclo celular.- Mayor expresión de IRS-2 y G6Pasa (almacenamiento de glucógeno)- Efecto antioxidante por aumento de la producción de ATP, metalotioneína y supresión de ERO- Diferenciación a linfocitos T productores de IL-22 | EsteatosisCirrosis | Tejido hepáticoR | Naseem et al.105 | ||
| IL-6 tiene una relación directa con el aumento de la proteína C reactiva, IL-4, IL-10 e IFN-ɣ.40 mg/día de prednisona favorecen la actividad hepatoprotectora de IL-6 hasta en un 21% de los pacientes tratados, debido a su aumento. | HA | SueroH | González-Reimers et al.77 | ||||
| Se reportan valores de IL-6 mayores en pacientes con cirrosis hepática vs. grupo control. | Cirrosis | Sangre periféricaH | Soresi et al.106 | ||||
| IL-8/CXCL8 | Células de KupfferCEH activadasHepatocitos dañados | Hepatocitos | CXCR1CXCR2 | Exacerba la acumulación de lípidos en los hepatocitos vía Akt/HIF-1αDisminuye la expresión de PPARα, reduciendo la β-oxidaciónInduce la activación de SREBP-1C, promoviendo la expresión de genes adipogénicos. | Esteatosis | Tejido hepáticoR | Wang et al.45 |
| Neutrófilos | Promueve la infiltración hepática de neutrófilos, activando las vías:- JAK2/STAT3 (proliferación)- PI3K/Akt (quimiotaxis)- MAPK (quimiotaxis)- Fosfolipasa C (PLC)/proteína cinasa C (PKC) (activación celular) | HA | Tejido hepáticoHNeutrófilosH | French et al.107Takami et al.108 | |||
| Monocitos | - Promueve la infiltración hepática de macrófagos. | HACirrosis | Tejido hepáticoH | Zimmermann et al.109 | |||
| Una mayor expresión de IL-8 en tejido hepático se asocia a un mal pronóstico e infiltración hepática de neutrófilosLa expresión intrahepática de IL-8 es un predictor de la mortalidad a 90 días. | HA | SueroHTejido hepáticoH | Domínguez et al.90 | ||||
| Niveles de IL-8 son altos en Child C y F4, aumentando conforme al grado de severidad y fibrosis | Cirrosis | SueroHTejido hepáticoH | Zimmermann et al.109 | ||||
| IL-10 | Células de KupfferLinfocitos T reguladores | Células de Kupffer | IL10R1IL10R2 | Efecto antiinflamatorioActivación de la arginasa en macrófagos con perfil inflamatorio (M1), promoviendo su apoptosis | Fibrosis (F2) | Tejido hepáticoR0,0H | Wan et al.101 |
| En pacientes con daño hepático se detectó mayor apoptosis de macrófagos M1 y mayor expresión de macrófagos M2 | EsteatosisFibrosis (F2) | Suero y tejido hepáticoH | Wan et al.101 | ||||
| IL-4 | Células de Kupffer | Linfocitos Th2 | IL-4R | Los niveles de IL-4 aumentan progresivamente a lo largo de 15 días después del ingreso de pacientes con HAPosiblemente debido al cambio de la respuesta de Th1 a Th2. | HA | SueroH | González-Reimers et al.77; González-Reimers et al.110 |
| Niveles bajos de IL-4 en pacientes cirróticos vs. no cirróticos y grupo control | Cirrosis | SueroH | González-Reimers et al.77 | ||||
| IL-13 | Linfocitos (Th2) | Miofibroblastos | IL-4Rα- IL13Rα1 | A través de la vía STAT6 estimula la síntesis de colágeno | Fibrosis | Tejido hepáticoR | Lee et al.111 |
| Células de Kupffer | Induce la producción de TGF-β1 | Fibrosis | Tejido hepáticoR | Lee et al.111 | |||
| Niveles séricos de IL-13 mayores en sujetos alcohólicos vs. no tomadoresMayores niveles en cirróticos vs. no cirróticos, aumentan conforme a la puntuación Child-Pugh | Cirrosis | SueroH | González-Reimers et al.77 | ||||
| IFN-ɣ | NKLinfocitos (Th1)NKT | Hepatocitos | IFNGR | Atenúa la esteatosis, disminuyendo la lipogénesis | Esteatosis | Tejido hepático y hepatocitosR | Cui et al.112 |
| Células de Kupffer | Promueve la activación de macrófagos M1, aumentando su producción de óxido nítrico y expresión de TNF-α | Esteatohepatitis | Monocitos/macrófagosR | Luo et al.113 | |||
| Hepatocitos | Activación de apoptosis vía JAK-STAT1-IRF3 | De todos los sujetos incluidos en el estudio:FibrosisF1 (48%),F2 (17%),F3 (18%)Cirrosis (17%) | Tejido hepáticoH | Stärkel et al.114 | |||
| CEH | Suprime la síntesis de colágeno por medio de Smad7 atenuando la señalización TGF-β/SMAD3 | Fibrosis (F1)* | Tejido hepáticoRCEH aisladas de tejido hepáticoR | Sun et al.115Jeong et al.116 | |||
| CEHNK | Induce directamente la apoptosis de CEH y detiene su ciclo celular. Mejora la citotoxicidad de las NK hacia las CEH de manera directa, por el mecanismo TRAIL y NKG2D. Mediante la señalización IFN-γ/STAT1 en las CEHEl alcohol funciona como inhibidor de esta víaEl alcohol induce una mayor expresión de TGF-β1 el cual inhibe a las NK mediante una regulación negativa de NKG2D, TRAIL e IFN- γ | Cirrosis | Tejido HepaticoR | Radaeva et al.117Lemmers et al.91Fertin et al.96 | |||
| Pacientes cirróticos presentaron niveles más altos vs. alcohólicos con consumo activo sin cirrosis y un grupo control | Alcohólicos sin EHACirrosis | SueroH | González-Reimers et al.77 | ||||
| IL-17 | Linfocitos (Th17) | NeutrófilosCEH | IL-17RA | Promueve la infiltración hepática de neutrófilosActiva JAK/STAT3; PI3K/Akt (proliferación y quimiotaxis de neutrófilos)Estimula la producción de IL-8 y CXCL1 en CEH | EsteatohepatitisHACirrosis | Tejido hepáticoH | Lemmers et al.91 |
| CEHCélulas de Kupffer | Promueve el proceso profibrogénicoProducción de colágeno tipo 1 en CEH por activación de STAT3Producción de TGF-β1 por las células de Kupffer por vía NF-κB. | Fibrosis (F1)* | Tejido hepáticoRCEH y células de Kuppfer aisladas de tejido hepáticoR | Meng et al.118 | |||
| Expresión más alta en fibrosis F4 vs. etapas intermedias e iniciales de fibrosis | HACirrosis | Tejido hepáticoH | Lemmers et al.91 | ||||
| TGF-β1 | Células de KupfferCEH | CEH | TGF-β1RTGF-β2R | TGF-β2R fosforila a TGF-β1R y se activa SMAD2/3 estimulando la síntesis de proteínas de matriz extracelular como el colágeno tipo i y ii e inhibiendo su degradaciónRegula negativamente a miARN-29, produciendo una regulación positiva de las proteínas de matriz extracelularInhibe la expresión de varias enzimas antioxidantes | Fibrosis (F1)* | Tejido hepáticoR | Jeong et al.56; Qu et al.119; Zhang et al.120 |
| NK | Regula negativamente la expresión de NKG2D, TRAIL e IFN-ɣ, afectando la citotoxicidad de las células NK hacia las CEH. | Fibrosis | Tejido hepáticoR | Jeong et al.56; Qu et al.119; Zhang et al.120 | |||
| MCP-1 o CCL2 | Células de KupfferHepatocitos | Monocitos circulantes | CCR2 | Promueve la infiltración hepática de monocitos circulantes | InflamaciónEsteatohepatitis | Monocitos circulantesR | Mandrekar et al.121 |
| Células de Kupffer | Inducción de la expresión de ARNm de citocinas proinflamatorias: TNF-α, IL-1β e IL-6 | Inflamación | Tejido hepáticoR | Mandrekar et al.121 | |||
| MonocitosCélulas endoteliales sinusoidales | Inducción de la expresión de ARNm de las moléculas de adhesión celular ICAM y del CD68. | EsteatohepatitisHA | Tejido hepáticoR | Mandrekar et al.121 | |||
| Hepatocitos | -Disminución en la expresión de ARNm de PPARα.-Disminución de la unión de PPARα al ADN.-Disminución del ARNm de ACC y CPT1 | Esteatosis | HepatocitosR | Mandrekar et al.121 | |||
| Concentraciones más altas en pacientes con HA vs. cirrosis y en sujetos control. | HA | SueroH | Fisher et al.122 |
ACC: acetil-CoA carboxilasa; ADN: ácido desoxirribonucleico; AKT: V-akt murine thymoma viral oncogene homolog; ARNm: ARN mensajero; ASC: proteína tipo punto asociada a apoptosis; ATP: adenosín trifosfato; BCL-1: B-cell lynphoma 1; BCL-2: B-cell lymphoma 2; CCR2: receptor tipo 2 de quimiocina C-C; CEH: células estelares hepáticas; CPT1: carnitina palmitoiltransferasa i; CXCL-8: interleucina 8; CXCR1: C-X-C motif chemokine receptor 1; CXCR2: C-X-C Motif Chemokine Receptor 2; EHA: enfermedad hepática alcohólica; ERO: especies reactivas de oxígeno; G6Pasa: glucosa-6-fosfatasa; HA: hepatitis alcohólica; HIF-1α: factor 1 inducible por hipoxia; ICAM: molécula de adhesión intercelular; IFN-γ: interferón gamma; IFNGR: receptor de interferón gamma; IL: interleucina; IL-1R1: receptor de interleucina tipo 1; IL-4R: receptor de interleucina 4; IL-4Rα/IL13Rα1: receptor de interleucina 4 alfa/ receptor de interleucina 13 alfa 1; IL-6R: receptor de interleucina 6; IL-6Ra: receptor de interleucina 6 alfa; IL-10R1: receptor de interleucina 10 tipo 1; IL-10R2: receptor de interleucina 10 tipo 2; IL-17RA: receptor de interleucina 17 A; IRF3: factor 3 regulador del interferón; IRS-2: sustrato del receptor de la insulina tipo 2; JAK: janus quinasa; MCP-1: proteína quimiotáctica de monocitos 1; MMP-9: metaloproteinasa 9; NF-kB: factor nuclear kappa B; NK: células natural killer; NKG2D: natural killer group 2D receptor; NKT: células natural killer T; NLRP3: proteína con dominio pirina 3 de la familia NLR; PI3K: fosfoinositol 3-cinasa; p38 MAPK: proteína cinasa activada por mitógenos p38; PPARα: receptor activado por proliferadores peroxisómicos tipo alfa; sIL-6Ra: receptor de IL-6 soluble; SMAD: familia Smad; SREBP1: proteína 1 de unión a elementos reguladores de esteroles; STAT3: transductor de señal y activador de la transcripción 3; STAT6: transductor de señal y activador de la transcripción 6; TGF-β1: factor de crecimiento transformante beta-1; TGF-β1R: receptor del factor de crecimiento transformante beta 1; TGF-β2R: receptor del factor de crecimiento transformante beta 2; TIMP1: inhibidor tisular 1 de la metaloproteinasa; TNFα: factor de necrosis tumoral α; TNFR1: receptor de necrosis tumoral; TRAIL: ligando inductor de la apoptosis relacionado con el factor de necrosis tumoral.
HInvestigación en humanos.
RInvestigación en ratones.
RaInvestigación en ratas.
R,HInvestigación en ratones y humanos.
Por su parte, la regulación celular también desempeña un papel preponderante en la EHA. Tal es el caso de las células NK, las cuales tienen un papel clave en la defensa del huésped contra la infección viral, la transformación tumoral y la inhibición de la fibrosis hepática; no obstante, contribuyen a la patogénesis de la inflamación y lesión hepática a través de su citotoxicidad natural descontrolada y producción de citocinas123,124. Las células NK activadas producen citocinas como IFN-γ, TNF-α, IL-10, IL-12, IL-22 y quimiocinas que incluyen MIP-1α, MIP-β, RANTES e IL-8125. A través de este mecanismo las células NK promueven la inflamación hepática, culminando en daño hepático. Este daño es debido en parte a la expresión de los receptores inhibidores y activadores en las células NK y de sus ligandos en hepatocitos y células no parenquimatosas, las cuales están significativamente alteradas durante la enfermedad hepática126, aumentando la actividad citotóxica y proinflamatoria de las NK contra dichas células, dando lugar a la eliminación de hepatocitos y, por lo tanto, a un mayor daño hepático127,128 (fig. 1K).
Regulación inmunológica durante la fibrosis hepáticaLa activación de las CEH es el evento clave de la fibrogénesis hepática1 (fig. 1). Se ha descrito que la inmunosupresión es un factor permisivo importante para el desarrollo de fibrosis, sugiriendo que el estado inmunológico del huésped regula la progresión de la fibrosis hepática117,129,130. Está bien documentado que en pacientes con abuso de alcohol e inmunodepresión la progresión de la fibrosis hepática es mucho más rápida117,129,130. Además, las CEH activadas producen inhibidores tisulares de metaloproteinasas, los cuales atenúan la actividad de las metaloproteinasas131,132. Adicionalmente, las CEH constituyen la fuente principal de TGF-β, siendo este último el inhibidor más potente de las funciones de las células NK a través de la regulación a la baja del receptor activador NKG2D56,123,133.
Las células NK y las Th1 producen IFN-γ, el cual induce directamente la apoptosis y el arresto del ciclo celular de las CEH116,124. Además, el IFN-γ potencia la actividad citotóxica de las células NK, lo que contribuye de manera significativa a su efecto antifibrótico117(tabla 1). El consumo de alcohol incrementa la actividad citotóxica de las células NK en individuos sin EHA, lo cual contribuye al desarrollo de la lesión hepática por alcohol134 (fig. 1K).
Actualmente, nuestro grupo de trabajo realiza estudios encaminados en los cambios y/o las alteraciones de las diferentes estirpes celulares de la respuesta inmunológica y de varios mediadores celulares a nivel periférico, con el objetivo de describir los cambios celulares y las vías de señalización que participan en los diferentes patrones de consumo, en la HA y en cirrosis hepática por alcohol.
ConclusiónLas citocinas son una pieza clave en la patogenia de la EHA, ya que dan lugar a la perpetuación del estado inflamatorio y al daño hepático. Así mismo, es importante enfatizar que la desregulación inmunológica es un proceso sistémico. No obstante, también son responsables de mecanismos hepatoprotectores para preservar la funcionalidad hepática, por lo que es de suma importancia conocer sus funciones en el desarrollo de la EHA, y generar intervenciones que frenen su progresión y mejoren el pronóstico. A pesar de clasificar la EHA en etapas estas no son estadios separados, ya que pueden coexistir en un mismo individuo. Es crucial que no se subestime el consumo de alcohol. El abuso y la dependencia no suelen ser diagnosticados, por lo que no hay posibilidad de frenar la evolución de la EHA.
PerspectivasLa EHA es altamente prevalente en México, por lo que deben tomarse medidas de prevención y tratamiento de manera apremiante. Aunque ya se han identificado moléculas que son útiles como blancos terapéuticos o marcadores de daño hepático, se necesita poner énfasis en los mediadores clave del estado patológico de los individuos con EHA. El entendimiento de los mecanismos implicados en la patogenia es el primer paso para el desarrollo de medidas terapéuticas que tengan un impacto en el curso de la enfermedad. Así mismo, el uso de marcadores séricos y otros métodos no invasivos podrían permitir una identificación de la enfermedad en etapas más tempranas, con el objetivo de tener esquemas de manejo de pacientes con EHA y poder reducir la morbilidad y la mortalidad, ya que hasta la fecha el trasplante es el único tratamiento efectivo, y con ello disminuir la carga económica que genera en el sistema de salud en el ámbito nacional.
Responsabilidades éticasEl presente trabajo es un manuscrito de revisión por lo que no involucra pacientes, animales de experimentación, ni se trata de un estudio clínico. Toda la información es citada de manera correcta, respetando así la autoría de cada trabajo. Adicionalmente, el grupo de trabajo y sus proyectos experimentales referentes a la enfermedad hepática alcohólica están apegados a la comisión de investigación y al comité de Ética del Hospital General de México Dr. Eduardo Liceaga y de la Facultad de Medicina de la UNAM.
FinanciaciónEl presente estudio fue parcialmente financiado por el Consejo Nacional de Ciencia y Tecnología (CONACyT) con número SALUD-2016-272579 y el Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT-UNAM) con número TA200515.
Conflicto de interesesLos autores no expresan ningún conflicto de intereses.

