




¿ Introduction
Hepatic encephalopathy (HE) is a neurological syndrome characterized by neuropsychiatric changes coupled to the presence of neuromuscular alterations resulting from severe progressive hepatic insufficiency, after exclusion of other known brain disease.1
Symptoms of HE include attention deficit, changes in sleep patterns and a lack of muscular coordination that can progress to stupor and coma.2 HE is classified into three distinct types: Type A, which occurs in patients with acute hepatic insufficiency; Type B, which arises in patients with portosystemic bypass; and Type C, when HE occurs in patients with cirrhosis.1 In addition, Type C HE is further subdivided into three subtypes:
A. Episodic: This subtype results from precipitating factors or can occur spontaneously in the absence of such factors. Therefore, the presence of gastrointestinal hemorrhage, uremia, infections, constipation, dehydration, hyponatremia, hypohyperkalemia, dietary transgressions, the use of diuretics, an increase in renal ammonium load, and the use of psychotropic drugs, among other factors, must be excluded (Table 1). When one or more episodes of HE occur within a single year, it is referred to as recurrent hepatic encephalopathy.

B. Persistent: This subtype includes cognitive deficits that negatively affect quality of life. The non-cognitive abnormalities, such as extrapyramidal disorders or sleep disturbances require separate analysis. This type of encephalopathy is subdivided, according to West Heaven classification, into mild HE (grade I HE) and severe (grades II to IV HE). Treatment-dependent persistent hepatic encephalopathy is a subgroup in which symptoms reappear quickly when treatment is suspended. The difference between persistent and episodic HE is that in the former, patients never become free of HE and in the latter patients remain asymptomatic between HE episodes (Figure 1).3,4
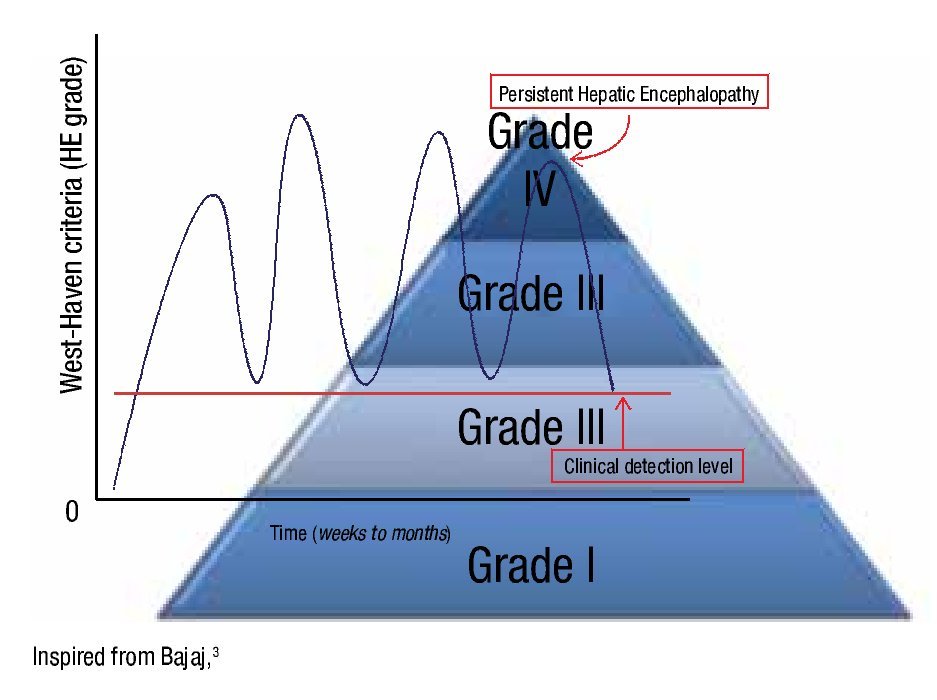
¿ Figure 1. Relationships between hepatic encephalopathy (HE) grades along time in persistent HE. These patients never become free of HE.
C. Minimal: Minimal HE (MHE) is identified when poor performance is observed in neuropsychological tests (e.g., number connection test or trail-making test/ digit symbol test), but the patient does not show obvious clinical changes. It is important to identify deterioration in cognitive function in patients with minimal HE, because it has been reported that these patients are at high risk for car accidents and possibly occupational accidents when handling machinery.5,6 The psychometric hepatic encephalopathy score (PHES), is a battery of neuropsychological tests, and recently has been validated for Mexican population. This set of tests helps in the diagnosis of MHE.7
Approximately 28% of cirrhotic patients develop HE during the course of the disease8 and sub-clinical neuropsychiatric changes have been identified in up to 84% of patients.9 This high prevalence coupled with the increasing incidence of chronic hepatic diseases (e.g., non-alcoholic steatohepatitis, hepatocarcinoma, etc.) and the longer survival associated with most effective treatment suggest that HE may become a highly frequent problem in clinical practice. In Mexico, an analysis of the trend in hepatic diseases has predicted that by the year 2050 there will be about 1.5 million chronic liver disease cases that will be susceptible to HE. Furthermore, it is known that cirrhosis primarily affects economically active population (35 to 55 years old), which may result in a significant economic burden.10 An accurate assessment of the pathophysiological basis of HE will facilitate better preventive and therapeutic approaches and will better assist patients with persistent HE, a problem commonly encountered in daily clinical practice. Therefore our aim was to review medical literature searching for evidence concerning persistent HE and precipitating factors in addition to classical ones.
¿ Methods
We identified the articles by searching PubMed (1966 to January 2011), EMBASE (1980 to January 2011) and Pubgle. Relevant articles with the terms persistent AND hepatic AND encephalopathy were selected. We extracted data on factors precipitating and perpetuating hepatic encephalopathy.
¿ Epidemiology of Type C chronic HE
There are few studies that have determined the prevalence of HE by examining an established cohort. Recently Romero-Gómez and collaborators in a cohort with 25% prevalence of minimal hepatic encephalopathy, reported an overt hepatic encephalopathy incidence of 18% to 44% at 29 months follow up, and linked to genetic factors.11 HE was reported in 13% of patients who have had a first episode of ascites12 and in more than 69% of patients hospitalized for variceal bleeding.13 Prevalence of minimal hepatic encephalopathy (MHE) depends on the methods used for its diagnosis, if psychometric tests and electroencephalograms are used, approximately 15% to 50% and 20%, are detected respectively.14-17
The mortality rate of each HE episode depends on the patient's degree of hepatic insufficiency and the severity of the episode. Interestingly, it has been reported that 42% and 23% of patients at 1 and 3 years, respectively, survive the first HE episode.18
Risk factors for the development of HE have not been completely identified. Portal hypertension with esophageal varices19 and advanced hepatic disease (Child C or ascites)18 have been identified as independent predictors of HE development. When transjugular intrahepatic portosystemic shunt (TIPS) is placed, older age (over 55 years old), previous history of HE, low hepatic pressure gradient, and a Child-Pugh score more than 11 are all considered independent predictors of the development of HE after shunt placement.20-23 In patients with persistent or recurrent HE, presence of spontaneous portosystemic shunts must be sought.24
Finally, the underlying causes of HE are identified in up to 97% of cases when HE is episodic, while it is only identified in 70% of the cases when HE is recurrent. Multiple precipitating factors of HE must be sought (Table 1).25,26
¿ Approach to persistent hepatic encephalopathy
Considering that persistent HE causes cognitive deficit with impact on quality of life, in addition to the usual treatment, there will always be a need to thoroughly investigate the causes that may result in persistent symptoms based on the following evidence:
A. Spontaneous Portosystemic Shunts: The use of transjugular intrahepatic portosystemic shunt (TIPS) and its relationship to HE development has been clearly established in patients with cirrhosis. Although the presence of a shunt would classify the disease as Type B HE, its persistence may perpetuate the chronic nature of HE. Patients possessing these shunts may present neuropsychological abnormalities and high levels of ammonium even in the absence of severe hepatic insufficiency.27 In patients with cirrhosis, the high incidence of HE after portal derivative surgery28 and the presence of radiological portocaval shunts29 has been confirmed; thus a relationship between the diameter of the anastomosis and the incidence of HE has been reported.30 Recently, Riggio and collaborators24 performed a case-control study founding that spontaneous shunts can perpetuate the chronicity of HE. In the aforementioned study, the suspected effect of spontaneous shunts was confirmed by conducting computerized tomography and three-dimensional reconstructions of the portal system and notably demonstrated that their presence is not directly related to the degree of hepatic insufficiency. However, CT showed preservation of the portal vein diameters in this group of patients, but increase in the diameters of the splenic and left renal veins, indicating that most of the flow is derived from the superior mesenteric vein and not from the portal vein, resulting in a shunt similar to surgical spleno-renal anastomosis.
In patients with changes in their neuropsychometric tests, brain spectroscopy was performed and revealed glutamine-glutamate peaks with a concurrent decrease in myo-inositol levels. These results were notably similar to the peaks found in patients with HE and cirrhosis who lacked the presence of shunts. Thus, it is thought that a spontaneous shunt can lead to HE even in the absence of severe hepatic damage.
Once identified, the shunts are treated with interventional radiology to reduce the shunt diameter with placement of coils or embolization, which results in a significant reduction of neurological symptoms. Rarely, there is a transient increase in portal pressure after the procedure that can result in ascites and esophageal varices, such problems must be treated under the guidelines already established.
B. Use of diuretics: In patients with cirrhosis, dehydration induced by the administration of diuretics is a common cause of HE, which is influenced by changes in volume. The mechanism by which HE is induced is not fully understood in this case, however, Jalan and collaborators31 recently demonstrated that plasma expansion significantly reduces the concentration of plasma ammonium, probably through an increased excretion of ammonium through the urine. This reduction in ammonium levels was reported in patients treated with albumin and was accompanied by a decrease in oxidative stress, which implies that the oxidative stress and ammonium may be linked to the pathogenesis of HE induced by diuretics, suggesting a possible role of albumin infusion as an effective treatment regimen.
It is probable that changes in ammonium homeostasis, secondary to volume expansion, increase the luminal flow by improving renal perfusion;32 in the same way, diuretics,33 are known to inhibit the transporters involved in the excretion of ammonium. Alternatively, this change in ammonium levels can also be achieved through a decrease in renal ammoniagenesis facilitated by a reduction in the production of angiotensin II, which is an important modulator of ammonium synthesis in the proximal tubule.34 The hypothesis proposed in the afore mentioned study by Jalan is supported by a direct correlation between the decrease in levels of angiotensin II and plasma ammonium, with results in an increase in the excretion of ammonium through urine, such as those patients with hepatorenal syndrome.35
Another important observation is that improvement in HE was largely achieved in patients who were treated with albumin when compared to patients treated with synthetic expanders (Gelofusine) despite the latter's ability to reduce the ammonium plasma concentrations. This implies that the mechanism by which albumin improves HE involves something other than an effect on volume expansion. It should be noted that albumin is a 66 kDa protein and constitutes 50% of the plasma protein content in healthy individuals. Among its numerous functions, albumin acts in the fixation of various substances such as fatty acids, bilirubins, bile salts, amino acids, and nitric oxide, reducing most of the sulfhydryl groups present at the extracellular level. As such, albumin can limit the production of reactive species and hence production of free radicals,36 improves the redox balance, and reduces oxidative stress.37 In the study performed by Jalan, this hypothesis was supported by the observed reduction in the levels of malondialdehyde in the group of patients who were administered albumin, a phenomenon that was not observed in the group administered synthetic expanders,31 suggesting that oxidative stress may be involved. This hypothesis was validated by the reduction in malondialdehyde levels and by evidence that oxidative stress is part of the physiopathology of HE, possibly via mitochondrial damage, involving the oxidation of membrane phospholipids and enzymes that facilitate energy metabolism.38 In fact, oxidative stress is a recently studied pathway that includes nitric oxide imbalance, endothelial dysfunction, messenger RNA oxidation, and down regulation of antioxidant pathways such as superoxide dismutase, catalase and glutathione peroxidase.39-42
C. Renal insufficiency: The cognitive compromise is a common problem observed in patients with cirrhosis, and it has multifactorial etiologic factors, considering that ammonium has a central role in its pathophysiology. Renal function, in addition, has proved to be an important factor in ammonium metabolism, so its association with cognitive changes may be related. Recently, Kalaitzakis and collaborators43 studied 128 cirrhotic patients and evaluated their cognitive dysfunction by performing neuropsychological tests. Of the 128 patients enrolled in the study, 41 (32%) demonstrated cognitive changes, and of these, 16 (13%) had renal insufficiency. When the group with renal insufficiency was compared to those individuals without renal insufficiency, the presence of cognitive changes was 69% vs. 31% (p = 0.001), respectively. Without inferring the etiology or severity of cirrhosis from these reported differences, we conclude that renal dysfunction in cirrhosis patients is implicated in the physiopathology of HE.
D. Anemia: It is well known that for many diseases, nutritional changes and metabolic or hormonal changes can alter cognitive function. Anemia caused by cerebral hypoxia has been shown to have a strong influence on cognition. However, the most well characterized group is perhaps those patients with chronic renal insufficiency where the concept of neuroprotection and/or neuroregeneration was established when the anemia is treated, specifically with erythropoietin. This treatment has not been studied in patients with hepatic damage, but understanding that erythropoietin acts as an antiapoptotic, antiinflammatory, antioxidant, neurotrophic and angiogenic factor, may invariably alter the therapeutic plan by modify the degree of oxidative stress via an alternative pathway in the pathophysiology of hepatic encephalopathy (Figure 2).44,45
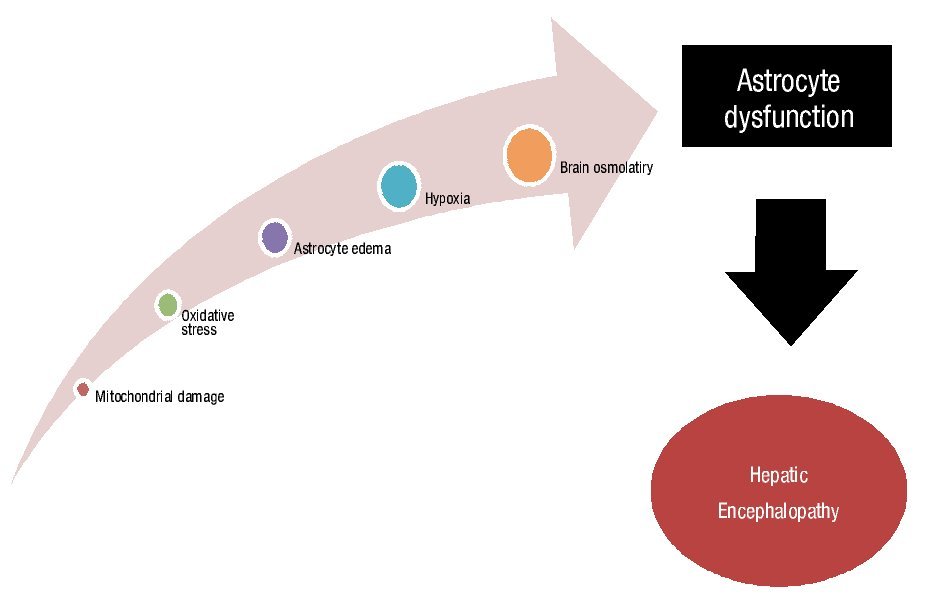
¿ Figure 2. Factors involved in astrocyte dysfunction and hepatic encephalopathy development and persistence.
E. Hydroelectrolytic changes: It has been shown that in patients with advanced cirrhosis there is a reduction in the concentration of several brain osmolytes, particularly myo-inositol. Dilutional hyponatremia significantly modifies the balance between these osmolytes, particularly glutamines and myo-inositol through an osmoregulatory mechanism to prevent the increase in the content of brain water, which can contribute to astrocyte dysfunction through the occurrence of HE (Figure 2). Restuccia and collaborators46 reported a decrease in myo-inositol levels and an increase in glutamine levels using spectroscopy and magnetic resonance imaging, indicating an increase in glutamine in relation to ammonium levels. In addition, the presence of low-grade cerebral edema was showed by measuring the magnetization transfer ratio (MTR), a magnetic resonance imaging modality that measures brain water content in cirrhotic compared with non-cirrhotic patients.47
F. Chronic gastrointestinal bleeding: The presence of acute intestinal bleeding is widely recognized in patients with HE, but recently the role of chronic bleeding has also been established in patients with HE. Romero-Gómez and collaborators observed that with an oral glutamine load (mimicking acute gastrointestinal bleeding and thus high ammonia load), patients had a higher risk of exhibiting HE, which has a significant impact on patient survival.48 Portal hypertensive gastropathy is found in 9% to 80% of cirrhotic patients, and is a cause of chronic gastrointestinal bleeding. Treatment consists in ethanol, aspirin and non-steroidal antiinflammatory drugs (NSAIDs) avoidance, beta-blockers therapy, TIPS or liver transplantation.49
G. Other factors perpetuating HE: Helicobacter pylori infection might contribute to persistent cognitive impairment in cirrhotic patients50,51 by increasing ammonia levels through urease activity. In this context, treatment of infected cirrhotic patients can help to diminish ammonia blood levels.
The main source of ammonia derives from small bowel gluthaminase activity and intestinal bacterium (85% of total body production). In a study of 34 cirrhotic patients, it was found HE in 85% of patients and 71% had a positive breath test for small intestinal bacterial overgrowth (SIBO). Prevalence of SIBO was 50%, 61% y 100% in patients without EH, grade I-II EH and grade III-IV EH, respectively (p = 0.04). This must be considered as a perpetuating factor and must be sought and treated when HE persists.52
¿ Treatment of persistent HE
Management of this entity must focus on search of all causes above mentioned, and treat as appropriated. Therapeutic options for HE are directed towards the reduction of ammonia production, increase in fixation and/or excretion of ammonia, direct neurological action and modifying porto-systemic collaterals.3,53 Therapies such as non-absorbable disaccharides (basically lactulose), antibiotics (rifaximin, neomycin, etc.), L-ornithine-L-aspartate, acetylcarnitine and acarbose may help to properly manage this clinical entity. Orthotopic liver transplantation constitutes the best therapy.3,54-58
¿ Conclusions
Persistent HE should always obligate to search for some factors that will be responsible for triggering and perpetuating this condition (Table 1). Several events can lead to HE, including oxidative stress, mitochondrial damage, persistent hypoxia, and changes in brain osmolarity resulting in astrocyte dysfunction with apparent HE (Figure 3). Understanding of pathophysiological issues, such those mentioned above, help to establish the best diagnostic and therapeutic approach in HE. Other non-classic factors may be involved in HE perpetuation, so anemia, spontaneous porto-systemic shunts, H. pylori infection, hydroelectrolytic imbalance and diuretic use must be excluded in these patients.
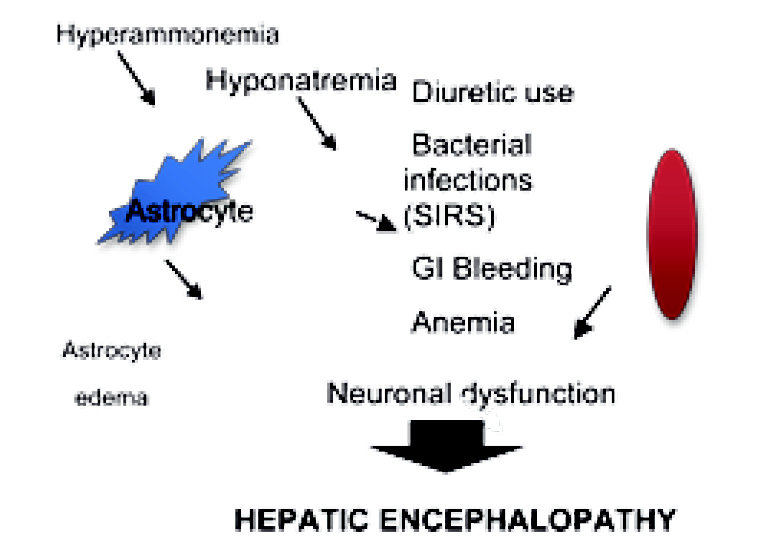
¿ Figure 3. Multiple factors affect the astrocyte, such as diuretic use, anemia and bacterial infections (systemic inflammatory response syndrome, SIRS) that give rise to hyponatremia and hyperammonemia. This in turn causes osmolyte reduction and eventually astrocyte edema. This edema is the cause of neuronal dysfunction that originates the clinical picture of hepatic encephalopathy.
¿ Acknowledgments
To the CONACYT (Consejo Nacional de Ciencia y Tecnología).
Correspondence author: Aldo Torre.
Vasco de Quiroga 15, Col. Sección XVI, Delegación Tlalpan, México D.F. C.P. 14000.
E-mail address: detoal@yahoo.com.
Received July 26th, 2011;
accepted September 20th, 2011.

