



¿ Introducción
El tumor desmoplásico de células pequeñas y redondas (TDCPR) es un tumor maligno muy poco frecuente y altamente agresivo que se caracteriza por afectar fundamentalmente a varones adolescentes. Fue descrito por primera vez en 1989 por Gerald y Rosai, recogiéndose en la bibliografía sólo unos pocos centenares de casos en todo el mundo.1-3
¿ Caso clínico
Hombre de 63 años que acudió al Servicio de Urgencias por dolor lumbar de nueve meses de evolución con diagnóstico previo de raquiestenosis medular; de intensidad creciente y que en los últimos cuatro meses se irradiaba a flanco y fosa ilíaca izquierda. Se acompañó de una pérdida de 20 kg de peso en el último año, astenia y anorexia. La exploración física mostró abdomen blando y depresible, doloroso de forma difusa, sobre todo en flanco y fosa ilíaca izquierda, sin signos de irritación peritoneal y sin masas palpables.
Los exámenes de laboratorio y marcadores tumorales (alfa-feto-proteína, antígeno carcinoembrionario, CA-125, CA-15-3, CA 19-9, antígeno prostático específico, beta2-microglobulina, gonadotropina coriónica humana beta, CYFRA 21.1), dentro de parámetros normales.
En la tomografía helicoidal computarizada (TC) abdominal se evidenció una masa retroperitoneal voluminosa de predominio izquierdo, de aproximadamente 13 cm en sus tres ejes, heterogénea, con centro necrótico, que englobaba la aorta y la vena cava inferior, sin poder definirse la vena renal izquierda y la esplénica, con circulación derivativa gástrica que infiltraba cuerpo y cola de páncreas, bazo, riñón izquierdo, psoas y cuerpo gástrico. Se observaron adenopatías retrocrurales y mediastinales junto con una tumoración suprarrenal derecha de aproximadamente 9 cm de eje mayor, heterogénea, que comprime el parénquima hepático (Figura 1).
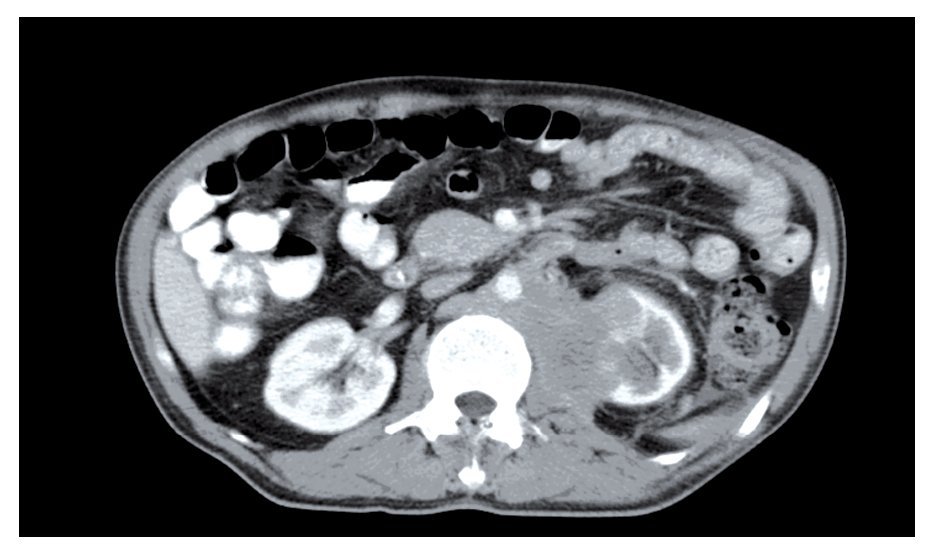
¿ Figura 1. Masa retroperitoneal de predominio izquierdo que compromete aorta y vena cava inferior y que infiltra cuerpo y cola de páncreas, bazo, riñón izquierdo, psoas y cuerpo gástrico.
La biopsia por punción (trucut) de la masa, mostró un área de infiltración por un tumor de células pequeñas con diferenciación neuroendocrina (Figura 2). El perfil de inmunohistoquímica es positivo para cromogranina y sinaptofisina, negativo para AE1, CD 45, CD 3, CD 79, CK 20, Leu 7 y desmina, CD 99 positivo/negativo, con un índice de proliferación celular (Ki 67) de 20% (Figura 3).
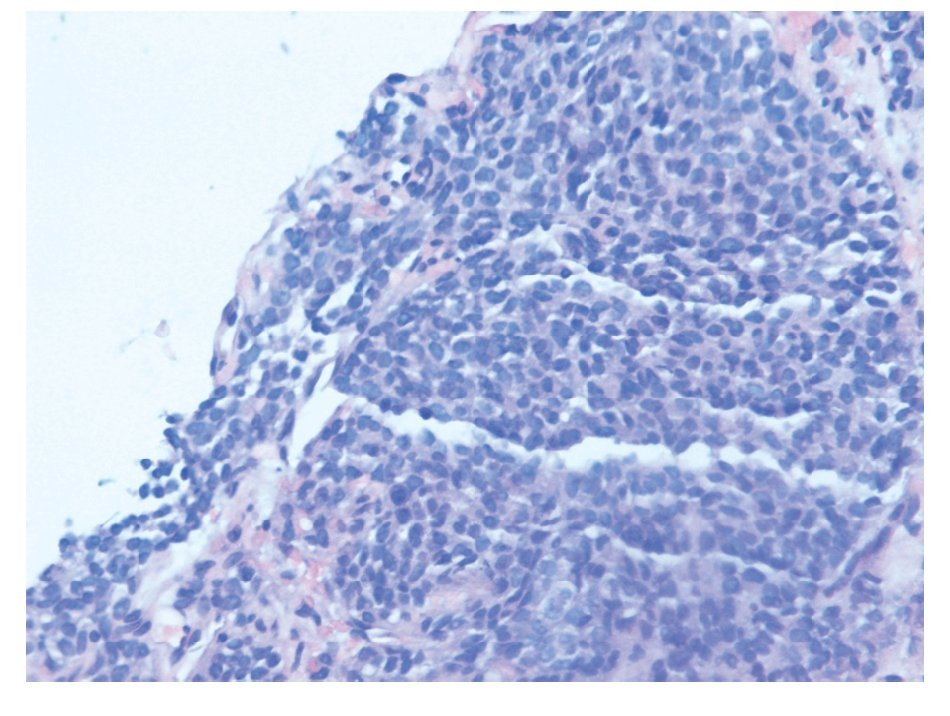
¿ Figura 2. Tinción con Hematoxilina- Eosina: nidos de células pequeñas y redondas con núcleo hipercromático envueltas en un estroma hipercromático hipocelular.
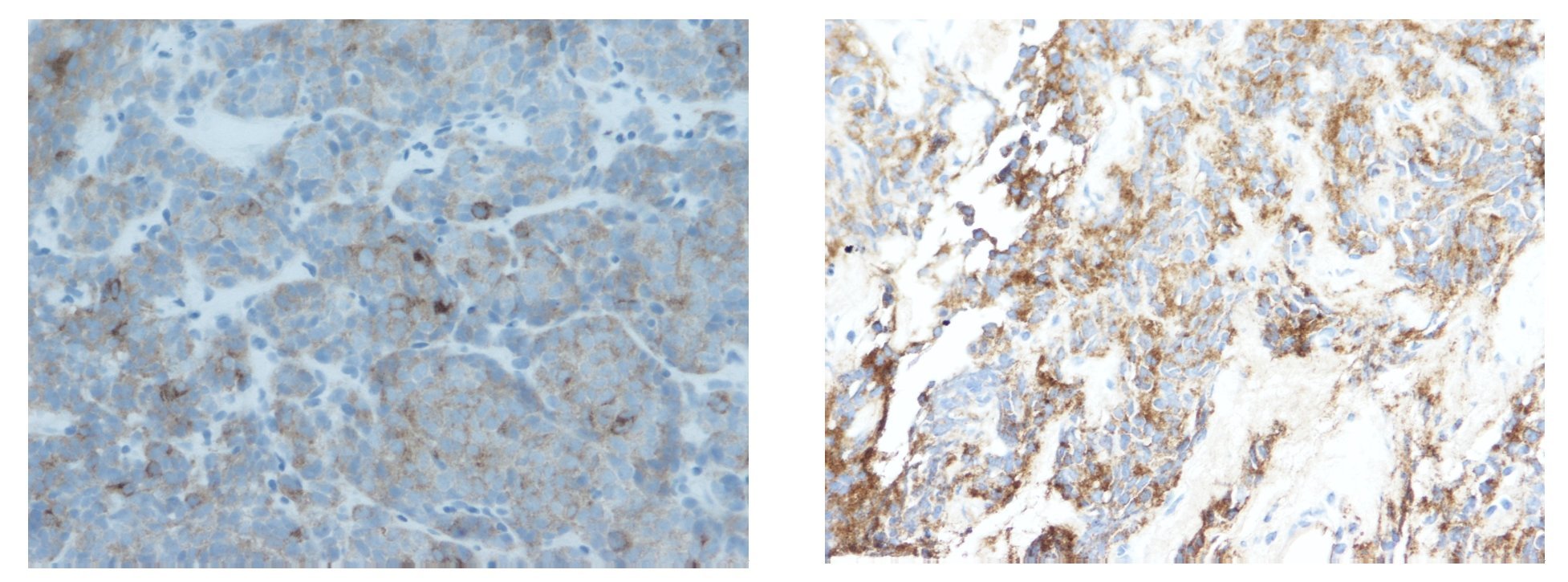
¿ Figura 3. Tinción positiva para cromogranina y sinaptofisina.
Debido a la imposibilidad para resecarlo, se decidió aplicar inicialmente un tratamiento quimioterápico paliativo según el esquema cisplatinoadriamicina-etopósido. Recibió cinco ciclos y posteriormente radioterapia pélvica por síndrome del psoas maligno, aunque con respuesta parcial. El paciente se mantenía vivo a los diez meses de ser diagnosticado.
¿ Discusión
El tumor desmoplásico de células pequeñas y redondas (TDCPR) es una neoplasia maligna infrecuente y muy agresiva. La localización más habitual del tumor esclerosante desmoplásico es la cavidad abdomino-pélvica, aunque se han descrito asimismo localizaciones paratesticulares, pleurales, en el hueso temporal, órbita, senos paranasales, ovario, entre otras.2,4 Su presentación clínica es inespecífica, consistiendo en la mayoría de los casos en dolor abdominal, masa palpable y síndrome constitucional, por lo que se necesita una prueba de imagen, generalmente la TC, para su detección.5 En el estudio histológico se visualizan nidos de células tumorales pequeñas, redondas y con núcleo hipercromático envueltas en un estroma hipercromático hipocelular. Los análisis de inmunohistoquímica son característicamente positivos para marcadores epiteliales (citoqueratina y antígeno epitelial de membrana), neurales (CD 56 y enolasa neuronal específica), musculares (desmina) y mesenquimales (vimentina) y para WT 1.6 La confirmación diagnóstica viene dada por la presencia de una traslocación cromosómica específica: t (11; 22), (p13; q12), documentada desde 1991, que origina la fusión del gen del tumor de Wilms y el gen del sarcoma de Ewing3,4,7 y que no ha podido ser demostrada en nuestro caso por la dificultad de acceso a las pruebas necesarias en nuestro hospital.
En la TC se presenta típicamente como una o múltiples masas de contornos lobulados, heterogéneas, con focos de necrosis y hemorragia, sin aparente órgano-dependencia. Es frecuente la presencia en el momento del diagnóstico de implantes peritoneales, metástasis hepáticas o adenopatías abdomino-pélvicas.4,5 No existen marcadores específicos, pero en 86% de los tumores intra-abdominales se eleva el CA 125, que se correlaciona con la existencia de ascitis, sin ser un buen marcador de la progresión de la enfermedad.4
El pronóstico de estos tumores es malo, con una supervivencia media entre 1.5 y 2.5 años.1 Debido a su rareza, no ha sido establecido por el momento un protocolo de tratamiento uniforme, aunque la terapia agresiva multimodal parece lo más recomendable. La resección quirúrgica completa es posible en raras ocasiones, debido a la frecuente presencia de metástasis, infiltración de las venas hepáticas o afectación diafragmática. No obstante, no parece aumentar de forma significativa la supervivencia por sí sola.1 La respuesta a la quimioterapia convencional es mala, habiéndose probado otras combinaciones, como el protocolo P6 (siete ciclos: uno, tres y seis: ciclofosfamida, doxorrubicina y vincristina, ciclos cuatro, cinco y siete: ifosfamida y etopósido) con mejores resultados. Por tanto, en este momento, en los tumores resecables se recomienda la combinación de quimioterapia con cirugía y radioterapia local, que ha mostrado los mejores resultados en términos de supervivencia en comparación con cada uno por separado. En los tumores no resecables, la quimioterapia paliativa es de dudosa indicación, si se valoran conjuntamente los beneficios y riesgos que ésta implica.7-9 De cara al futuro, se están estudiando otras alternativas, como el soporte con células madre autólogas, los anticuerpos monoclonales frente a los antígenos G (D2) y 8H9, los antiandrógenos y la perfusión continua peritoneal hipertérmica.6,10
En conclusión, los TDCPR son tumores raros y poco estudiados, pero que deben ser tenidos en cuenta en el diagnóstico diferencial de tumores intra-abdominales agresivos, pudiendo presentarse a cualquier edad y dar manifestaciones clínicas muy variadas, como de hecho sucedió en nuestro caso.
Correspondencia: Dra. Celia Camarero Mulas.
Calle Alcalá 160, piso 4º A, C.P. 28028, Madrid, España.
Teléfono: (+34) 699315795.
Correo electrónico: zelyakam@yahoo.es
Recibido el 19 de marzo de 2010;
aceptado el 23 de abril de 2010.

