




La Tríada de Currarino es un trastorno hereditario poco frecuente que pertenece al grupo de las malformaciones neuroentéricas y se caracteriza por malformación ano-rectal, defecto sacrococcígeo y tumor presacro que en la mayoría de las ocasiones corresponde a un teratoma o mielomeningocele anterior.1-3 La malformación ano-rectal más común es la estenosis o atresia anal. El defecto sacrococcígeo más frecuente corresponde a hemisacro o sacro en forma de cimitarra, sin embargo, pueden existir otras anormalidades segmentarias. La incidencia real de este síndrome se desconoce, existen menos de 250 casos publicados en la literatura mundial y se cree que muchos casos pasan desapercibidos, principalmente por la variabilidad fenotípica intra e interfamiliar.4-7 La sintomatología es variable entre los pacientes pero el síntoma más importante es el estreñimiento crónico desde el nacimiento.3,8 Presentamos el caso de un paciente con SC con expresión fenotípica completa.
Presentación del casoSe trata de un niño de dos años de edad con estreñimiento desde el nacimiento tratado con múltiples laxantes. A los cuatro meses de edad se hizo el diagnóstico de malformación ano-rectal tipo fístula recto perineal por lo que se le realizó una anoplastia radiada. Su evolución posoperatoria fue con estreñimiento y distensión abdominal requiriendo manejo con enemas. A los nueve meses de edad fue valorado por un cuadro de obstrucción intestinal baja, durante la exploración física el ano estaba permeable y un dilatador Hegar número 15 se introdujo sin dificultad. Al tacto rectal se encontró un tumor de consistencia blanda a nivel del promontorio. Posterior a la desimpactación fecal e irrigaciones del colon con una sonda transrectal multiperforada (colostomía intubada) se realizaron placas AP y laterales de columna lumbosacra, ultrasonido renal, y un colon por enema con hidrosoluble. La radiografía de columna demostró la presencia de un hemisacro (figura 1), el colón por enema evidenció la presencia de un mega-rectosigmoides (figura 2) mientras que el ultrasonido renal fue normal. Por lo anterior, se realizó una resonancia magnética que demostró una agenesia parcial de sacro y un tumor presacro (figura 3). Se encontraron raíces nerviosas lumbosacras con médula espinal a nivel de L3 y dilatación en canal central ependimario constituyendo una hidromielia secundaria. El paciente fue sometido a resección del megarectosigmoides con cierre del recto en bolsa de Hartmann y colostomía terminal en el colon descendente. Un mes después, se realizó un abordaje sagital posterior para exponer la región presacra, se identificó una lesión entre el recto y sacro que fue resecada en un 100% (figura 4). En el estudio histopatológico del segmento intestinal se identi- ficaron plexos nerviosos submucoso y mientérico con células ganglionares presentes. El estudio histopatológico de la neoplasia presacra reveló la presencia de tejido adiposo, fibras de músculo liso, glándulas revestidas por epitelio cilíndrico de tipo respiratorio y glia. Con estos hallazgos se hizo el diagnóstico de un teratoma maduro. A tres años de seguimiento, el paciente se encuentra bien sin estreñimiento residual y con control de esfínteres normal.
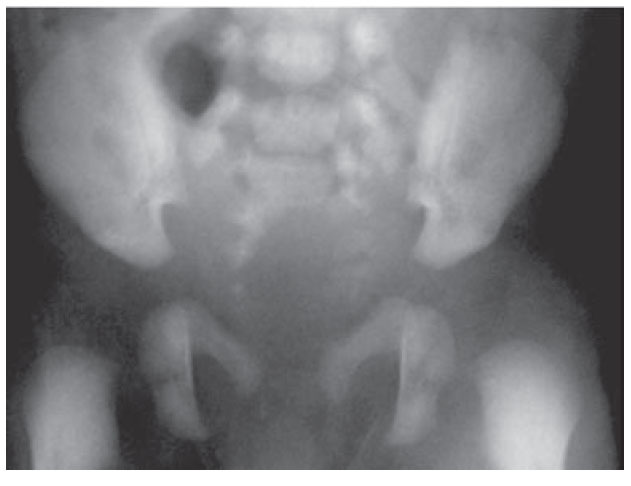
Figura 1. Hemisacro (Radiografía de columna dorsal).
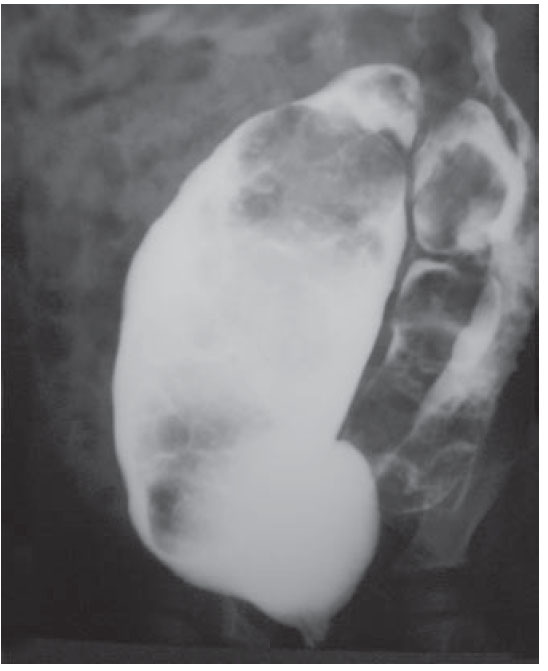
Figura 2. Colon por enema con mega-rectosigmoides.
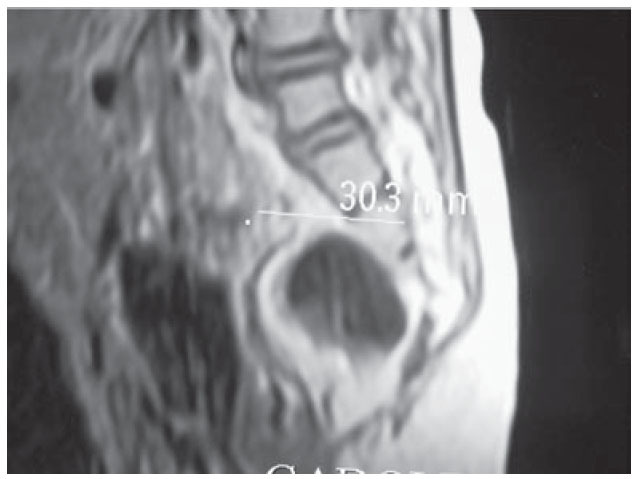
Figura 3. Agenesia parcial del sacro y lesión sacular en región anterior sacra (IRM).
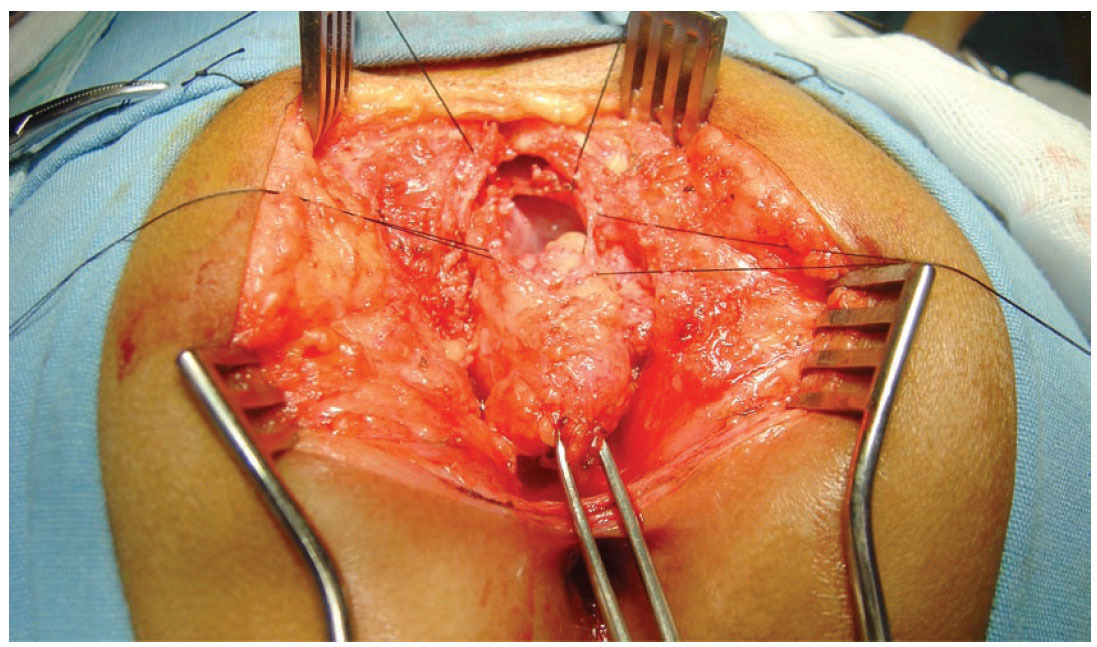
Figura 4. Resección quirúrgica del tumor.
DiscusiónEn 1926, Kennedy describió por primera vez la tríada de malformación ano-rectal, deformidades en el sacro y tumor presacro,9 sin embargo, fue hasta 1981 que Currarino y cols. reconocieron la combinación de estas alteraciones como un síndrome con una embriogénesis en común.1 Existe debate entre las teorías de la embriogénesis, Currarino y cols. propusieron una explicación simplificada para estas anormalidades basados en el síndrome notocordal dividido. Éste es un mecanismo en el que adherencias endoectodérmicas y defectos en la notocorda durante la vida fetal temprana resultan en una fístula entre el intestino y el canal espinal con presencia ventralmente de elementos entéricos y dorsalmente de elementos neurogénicos. La combinación de endodermo, ectodermo y mesodermo puede resultar en un teratoma, meningocele, quiste entérico o combinación de estos dos últimos. La malformación ano-rectal puede ser resultado simplemente de las adherencias descritas. Otra de las teorías más aceptadas es la de Gegg y colaboradores, quienes señalan qué errores en la migración de la eminencia caudal después de la neurulación puede causar las alteraciones presentes en el SC.5
Genéticamente se han descrito, en los últimos años, mutaciones en el gen HLXB9 localizado en el cromosoma 7 (región 7q36) como responsables del SC.4,10,11 La expresión fenotípica de las mutaciones de este gen es muy variable y va desde pacientes asintomáticos hasta pacientes con la tríada completa. En el 60% de los casos existe una historia familiar.12,13 Este síndrome se diagnostica en más de 80% de los casos en la primera década de vida mientras que en la forma incompleta, el diagnóstico se hace predominantemente en adultos.14,15
En pacientes pediátricos, el diagnóstico se debe sospechar en niños con estreñimiento crónico después del nacimiento con y sin malformación ano-rectal. Se debe realizar una exploración física adecuada identificando en primera instancia el tipo de malformación ano-rectal, el examen digital del recto localiza la estenosis rectal distal y puede palpar en algunos casos el tumor presacro. Las radiografías AP, lateral y USG de sacro son útiles para mostrar el defecto óseo sacro y la presencia del tumor, sobre todo, en recién nacidos o lactantes. La TAC y RM son los estudios de imagen de elección para confirmar el diagnóstico y para especificar las anormalidades de la médula espinal.16,17
La malformación ano-rectal más frecuente es la estenosis ano-rectal, seguido de la estenosis rectal, atresia y ectopia anal con o sin fístula y duplicación intestinal.18 El defecto óseo en la mayoría de los casos es un sacro en cimitarra o en forma de hoz, sin embargo, puede variar desde una desviación coccígea lateral hasta una ausencia del segmento sacro distal.3 El tumor presacro es un mielomeningocele en el 60% de los casos, teratoma benigno en 23%, quiste dermoide 6%, tumor no clasificado 4%, lipoma 2%, neurofi- bromatosis 1%, quiste entérico 1% y hamartoma 1%.8 En la literatura existe un informe de seis pacientes con SC asociado con neoplasias malignas, en dos niños y cuatro adultos. Los diagnósticos fueron teratoma maligno en cuatro casos, leiomiosarcoma en un caso y tumor neuroendocrino en un caso.13,19,20
El síntoma más común es el estreñimiento crónico desde el nacimiento, que en la mayoría de los casos se debe a compresión extrínseca del tumor presacro, estenosis anal o a la presencia de médula espinal anclada. El cuadro clínico puede simular una enfermedad de Hirschsprung y requerir biopsia rectal. En nuestro caso, la causa principal de constipación fue atribuida a la compresión extrínseca del tumor. Otros de los síntomas descritos son infecciones perianales recurrentes, meningitis, náusea, cefalea, dolor de espalda y reflujo vesicoureteral. El tratamiento quirúrgico de los pacientes con SC está enfocado a resolver el tipo de malformación ano-rectal y resecar el tumor presacro. La anorrectoplastia sagital posterior es un procedimiento bien establecido para este tipo de pacientes, sin embargo, cuando la médula anclada es sintomática se sugiere la vía perineal y transacra sagital posterior que permite el tratamiento simultáneo del intestino, tumor presacro y anormalidades neurológicas. Algunos autores señalan que en caso de existir cóccix, el cirujano debe resecar el tumor presacro junto con este hueso por la posibilidad de trasformación maligna del tumor y esto es más fácil por vía perineal transacra sagital posterior. 21,22
La incidencia del SC se desconoce, existen alrededor de 250 casos publicados en la literatura mundial y hasta donde sabemos éste es el primer caso de SC informado en México. La incidencia real seguramente es mucho más alta debido a que muchos pacientes pueden permanecer sin diagnóstico, esto gracias a que la expresión fenotípica de este síndrome en ocasiones es incompleta y los pacientes pueden cursar únicamente con estreñimiento crónico y defecto sacro asintomático. El avance en las técnicas de neuroimagen, en especial la RM ha ayudado no sólo a reconocer más casos de SC sino también a planificar y establecer una estrategia quirúrgica en pacientes con mielomeningocele, siringomielia, cono medular bajo o médula espinal anclada.
ConclusiónEl abordaje diagnóstico de pacientes con estreñimiento crónico en edad pediátrica y adulta debe considerar al SC como un diagnóstico diferencial cuyo manejo y tratamiento debe realizarse de manera multidisciplinaria sin olvidar que un diagnóstico y tratamiento temprano son esenciales. El estudio de otros miembros de la familia, particularmente niños deben ser evaluados tomando en cuenta el modo de herencia autosómico dominante. Finalmente, en todo paciente pediátrico con malformación ano-rectal deben realizarse estudios de imagen para descartar un SC.
Correspondencia: Dr. Roberto Vargas González. Laboratorio de Inmunopatologia de Puebla. Calle 16 sur 3902 Int. 102B Colonia Anzures, Puebla, México. CP. 72190 Tel: (52) (222) 4-03-50-23 Fax: (52) (222) 4-03-50-24 e-mail: dr.robertovargas@inmunopat.com
Fecha recibido: 10 marzo 2008 • Fecha aprobado: 27 julio 2008

