


La malabsorción congénita de glucosa y galactosa (MCGG) es un trastorno autosómico recesivo infrecuente, producido por la alteración del cotransportador de sodio-glucosa tipo 1 (SGLT1), ubicado en el borde en cepillo del intestino delgado, encargado de absorber glucosa y galactosa desde la luz intestinal hacia el enterocito, impulsados por un gradiente electroquímico transmembrana de sodio (fig. 1). La deficiencia congénita de SGLT1 es secundaria a mutaciones del gen SLC5A1, localizado en el cromosoma 22q13.11,2.
 Transportador SGLT 1: realiza el transporte activo en el enterocito de glucosa y galactosa contra un gradiente de concentración desde la luz intestinal hacia el enterocito. La energía requerida para hacer posible este transporte la adquiere del cotransporte de sodio en la misma dirección; b) ausencia del transportador SGLT 1: Al no absorberse glucosa ni galactosa, estos azúcares permanecen en la luz intestinal y arrastran agua por efecto osmótico, conduciendo a diarrea osmótica y deshidratación grave3. El resultado neto es una pérdida mayor de agua que de sodio, lo que concentra el sodio plasmático, llevando a hipernatremia.")
a) Transportador SGLT 1: realiza el transporte activo en el enterocito de glucosa y galactosa contra un gradiente de concentración desde la luz intestinal hacia el enterocito. La energía requerida para hacer posible este transporte la adquiere del cotransporte de sodio en la misma dirección; b) ausencia del transportador SGLT 1: Al no absorberse glucosa ni galactosa, estos azúcares permanecen en la luz intestinal y arrastran agua por efecto osmótico, conduciendo a diarrea osmótica y deshidratación grave3. El resultado neto es una pérdida mayor de agua que de sodio, lo que concentra el sodio plasmático, llevando a hipernatremia.
Hasta la fecha se han reportado aproximadamente 300 casos en el mundo3. Requiere un alto índice de sospecha, pudiendo ser potencialmente mortal si no se realiza un diagnóstico oportuno.
Presentamos a una lactante de 44 días de edad, fruto de la segunda gestación de una madre sin controles prenatales y con antecedente de consanguinidad. Nació de 37 semanas, por cesárea, con un peso 2,865g y una talla de 49cm. A los 10minutos de vida presentó dificultad respiratoria, posteriormente distensión abdominal y drenaje bilioso por sonda. Sospecharon sepsis neonatal temprana y enterocolitis necrosante, e indicaron antibióticos y ayuno por 5días. Los estudios infecciosos realizados resultaron negativos. Posteriormente se inició lactancia materna con aparición de deposiciones líquidas, hasta 10episodios al día, condicionando una deshidratación severa, intolerancia a la vía oral y pérdida severa de peso (13.4%). Se sospechó alergia a la proteína de la leche de vaca y se manejó con dieta de restricción a la madre; posteriormente se introdujo en su dieta fórmula extensamente hidrolizada y elemental, persistiendo el vómito y la diarrea de alto gasto, con acidosis metabólica hiperclorémica e hipernatremia, sin respuesta a la corrección con agua libre, por lo que remiten.
Examen físico de ingreso: fontanela deprimida, abdomen distendido, peristaltismo aumentado. Antropometría: Peso: 2.800g (P/T: –1.35DE). Talla 49cm (T/E: –3.41DE). Se registró tendencia a la poliuria. Paraclínicos: acidosis metabólica con anión gap elevado, hipernatremia, hipercloremia y azoados elevados (tabla 1). Radiografía de abdomen simple: niveles hidroaéreos. Ecografía de abdomen: moderada distensión de asas intestinales. Se sospechó una MCGG. Se colocó sonda vesical, evidenciando que las pérdidas eran predominantemente gastrointestinales. Se indicó ayuno, con nutrición parenteral total, observando mejoría de gasto fecal. Se inició una fórmula artesanal a base de fructosa, proteína y aceite de oliva, asociada a multivitaminas y oligoelementos (tabla 2). Toleró volúmenes progresivos de nutrición enteral, con lo que resolvió sus síntomas gastrointestinales.
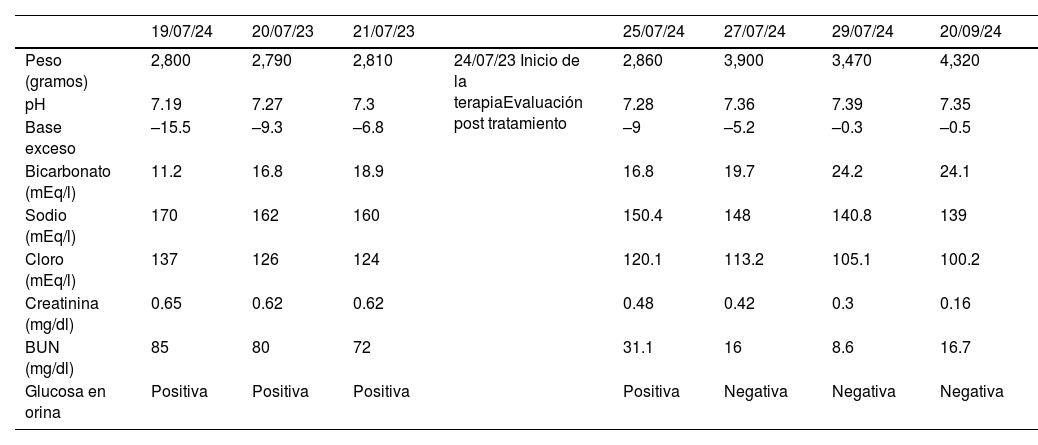
Evolución ponderal y de exámenes de laboratorio de la paciente
| 19/07/24 | 20/07/23 | 21/07/23 | 25/07/24 | 27/07/24 | 29/07/24 | 20/09/24 | ||
|---|---|---|---|---|---|---|---|---|
| Peso (gramos) | 2,800 | 2,790 | 2,810 | 24/07/23 Inicio de la terapiaEvaluación post tratamiento | 2,860 | 3,900 | 3,470 | 4,320 |
| pH | 7.19 | 7.27 | 7.3 | 7.28 | 7.36 | 7.39 | 7.35 | |
| Base exceso | –15.5 | –9.3 | –6.8 | –9 | –5.2 | –0.3 | –0.5 | |
| Bicarbonato (mEq/l) | 11.2 | 16.8 | 18.9 | 16.8 | 19.7 | 24.2 | 24.1 | |
| Sodio (mEq/l) | 170 | 162 | 160 | 150.4 | 148 | 140.8 | 139 | |
| Cloro (mEq/l) | 137 | 126 | 124 | 120.1 | 113.2 | 105.1 | 100.2 | |
| Creatinina (mg/dl) | 0.65 | 0.62 | 0.62 | 0.48 | 0.42 | 0.3 | 0.16 | |
| BUN (mg/dl) | 85 | 80 | 72 | 31.1 | 16 | 8.6 | 16.7 | |
| Glucosa en orina | Positiva | Positiva | Positiva | Positiva | Negativa | Negativa | Negativa |
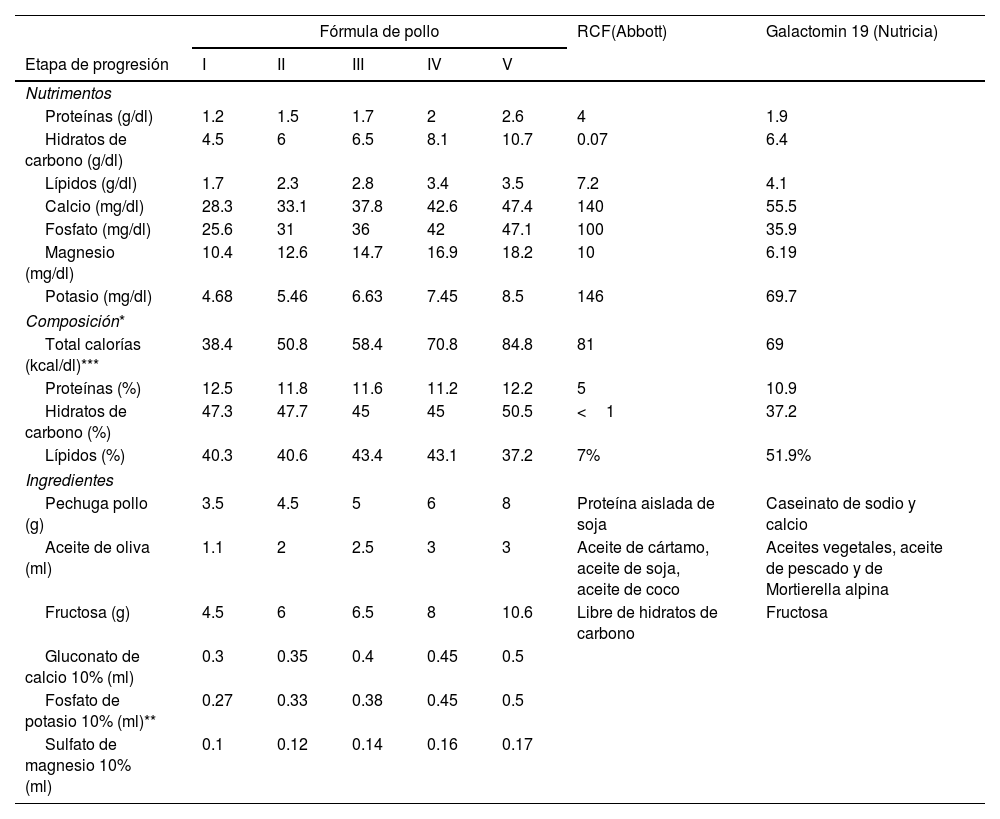
Protocolo de introducción progresiva de la fórmula artesanal a base de fructosa
| Fórmula de pollo | RCF(Abbott) | Galactomin 19 (Nutricia) | |||||
|---|---|---|---|---|---|---|---|
| Etapa de progresión | I | II | III | IV | V | ||
| Nutrimentos | |||||||
| Proteínas (g/dl) | 1.2 | 1.5 | 1.7 | 2 | 2.6 | 4 | 1.9 |
| Hidratos de carbono (g/dl) | 4.5 | 6 | 6.5 | 8.1 | 10.7 | 0.07 | 6.4 |
| Lípidos (g/dl) | 1.7 | 2.3 | 2.8 | 3.4 | 3.5 | 7.2 | 4.1 |
| Calcio (mg/dl) | 28.3 | 33.1 | 37.8 | 42.6 | 47.4 | 140 | 55.5 |
| Fosfato (mg/dl) | 25.6 | 31 | 36 | 42 | 47.1 | 100 | 35.9 |
| Magnesio (mg/dl) | 10.4 | 12.6 | 14.7 | 16.9 | 18.2 | 10 | 6.19 |
| Potasio (mg/dl) | 4.68 | 5.46 | 6.63 | 7.45 | 8.5 | 146 | 69.7 |
| Composición* | |||||||
| Total calorías (kcal/dl)*** | 38.4 | 50.8 | 58.4 | 70.8 | 84.8 | 81 | 69 |
| Proteínas (%) | 12.5 | 11.8 | 11.6 | 11.2 | 12.2 | 5 | 10.9 |
| Hidratos de carbono (%) | 47.3 | 47.7 | 45 | 45 | 50.5 | <1 | 37.2 |
| Lípidos (%) | 40.3 | 40.6 | 43.4 | 43.1 | 37.2 | 7% | 51.9% |
| Ingredientes | |||||||
| Pechuga pollo (g) | 3.5 | 4.5 | 5 | 6 | 8 | Proteína aislada de soja | Caseinato de sodio y calcio |
| Aceite de oliva (ml) | 1.1 | 2 | 2.5 | 3 | 3 | Aceite de cártamo, aceite de soja, aceite de coco | Aceites vegetales, aceite de pescado y de Mortierella alpina |
| Fructosa (g) | 4.5 | 6 | 6.5 | 8 | 10.6 | Libre de hidratos de carbono | Fructosa |
| Gluconato de calcio 10% (ml) | 0.3 | 0.35 | 0.4 | 0.45 | 0.5 | ||
| Fosfato de potasio 10% (ml)** | 0.27 | 0.33 | 0.38 | 0.45 | 0.5 | ||
| Sulfato de magnesio 10% (ml) | 0.1 | 0.12 | 0.14 | 0.16 | 0.17 | ||
Ingredientes y composición por 100ml y comparación de información nutricional con las fórmulas comerciales a base de fructosa no disponibles en Colombia.
Control de exámenes de laboratorio: mejoría de la acidosis metabólica, anión gap, hipernatremia, azoados y tasa de filtración glomerular. Se descartó tubulopatía. El estudio genético confirmó una mutación en el gen SLC5A1 c.1673 G>A (p.Arg558His) en estado homocigoto.
A las 2 semanas de manejo nutricional presentó recuperación del peso (3,470g) y mejoría de la diarrea. La paciente egresó con restricción de glucosa y galactosa.
Actualmente tiene 16 meses, tolera la fórmula artesanal establecida asociada a la complementaria, con restricción de alimentos ricos en glucosa y galactosa, con deposiciones consistentes, 2 veces día, con ganancia pondoestatural, paraclínicos con electrolitos normales, Sudán III negativo, función renal estable y hemograma normal.
La diarrea osmótica de alto gasto en la MCGG es debida a la malabsorción de los hidratos de carbono (glucosa y galactosa) que, al no ser absorbidos en el intestino delgado, son fermentados por las bacterias colónicas, produciendo ácidos grasos de cadena corta que generan heces acuosas y ácidas (pH fecal<5,3), que pueden llegar a confundirse con orina4, como sucedió en nuestra paciente. Esta diarrea conlleva deshidratación hipernatrémica, acidosis metabólica, pérdida de peso, distensión abdominal, cólico, vómito, poliuria, nefrolitiasis, nefrocalcinosis y deterioro de la función renal debido a la deshidratación crónica5. El tiempo promedio al diagnóstico es de aproximadamente 8 semanas, pero puede tardar hasta 4.5meses6. El diagnóstico se inicia con la sospecha clínica; la eliminación de glucosa y galactosa de la dieta resuelve la diarrea casi de inmediato y corrige la hipernatremia, por lo que estos pacientes responden favorablemente al ayuno7. Los criterios diagnósticos están descritos en la figura 2. El estudio de mutaciones del gen SLC5A1 confirma el diagnóstico8. Se ha descrito la mutación genética SLC5A1:p.Arg558His/c.1673G>A en homocigosis causante de MCGG3. En nuestra paciente el diagnóstico clínico se estableció a las 7 semanas de vida y se basó en la asociación de diarrea crónica e hipernatremia persistente. La alteración electrolítica esperable en diarrea es la hiponatremia. La presencia de hipernatremia persistente en diarrea de alto gasto a temprana edad orienta a pensar en una MCGG. Gracias a que se sospechó oportunamente, se pudo iniciar un manejo nutricional empírico que rápidamente controló los síntomas de la paciente, mientras se obtuvo el estudio genético.

El tratamiento es nutricional, a base de fórmulas con fructosa, debido a que este monosacárido tiene un transportador diferente al SGLT-1, denominado GLUT5. En el mercado existen 2 fórmulas: RCF (Abbott Nutrition) y Galactomin 19 (Nutricia)9. Sin embargo, en Colombia no se comercializan, por lo que nuestra paciente recibió una fórmula artesanal a base de fructosa (tabla 2), lo que permitió la resolución de los síntomas. Con el manejo nutricional el pronóstico es bueno; a corto y mediano plazo pueden tolerar pequeñas cantidades de glucosa, incluso a largo plazo mayores cantidades sin presentar diarrea10.
En conclusión, el reconocimiento oportuno de MCGG permite realizar un adecuado manejo, disminuyendo la morbimortalidad de la enfermedad. Hasta donde conocemos este es el primer caso reportado en Colombia con confirmación genética de la mutación c.1673 G>A (p. Arg558His) en el gen SLC5A1. Este hallazgo constituye un punto de partida para futuros estudios que evalúen la prevalencia de mutaciones de este gen en Latinoamérica y sus implicaciones en consejería genética.
Responsabilidades éticasLos autores declaramos que para la realización de este artículo se cumplió el derecho a la privacidad y no aparecen datos de pacientes.
Para la confidencialidad de datos se han seguido los protocolos de nuestra institución sobre la publicación de datos de pacientes. En el presente trabajo no fue necesario autorización del comité de ética por tratarse de una descripción en retrospectivo de la evolución de un caso clínico, el cual cuenta con el consentimiento informado y autorización de los padres del paciente para su publicación.
Declaramos que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
FinanciaciónNo se recibió patrocinio de ningún tipo para llevar a cabo este artículo.
Conflicto de interesesLos autores declaramos no tener ningún conflicto de intereses.

