


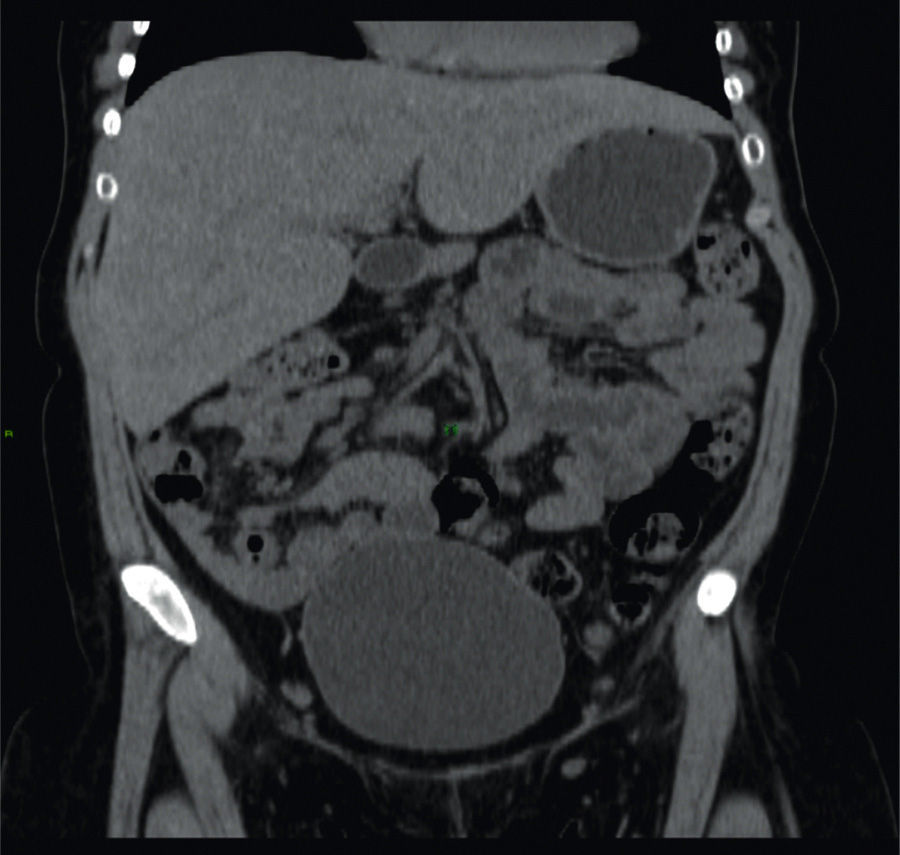
A 26-year-old woman was diagnosed with type 1 diabetes mellitus (DM1) at the age of 5 years. Her disease was chronically uncontrolled despite multiple insulin regimens, with HbA1c levels of 12%, and she presented with retinopathy and peripheral neuropathy. Clinical symptoms of one-month duration were characterized by nausea, vomiting, and abdominal pain. Physical examination revealed very short stature, overweight, Cushingoid facies, and hepatomegaly. Liver function tests only showed increased transaminases (ALT 158 IU, AST 192 IU), and AF 265 IU. Viral or autoimmune etiology was ruled out. A noncontrast abdominal computed tomography (CT) scan revealed hepatomegaly with a shiny aspect (fig. 1).
Percutaneous liver biopsy revealed normal architecture with mild changes due to fat infiltration (fig. 2A). Periodic acid-Schiff (PAS) stain showed abundant glycogen deposits (fig. 2B) that disappeared after diastase digestion (fig. 2C).
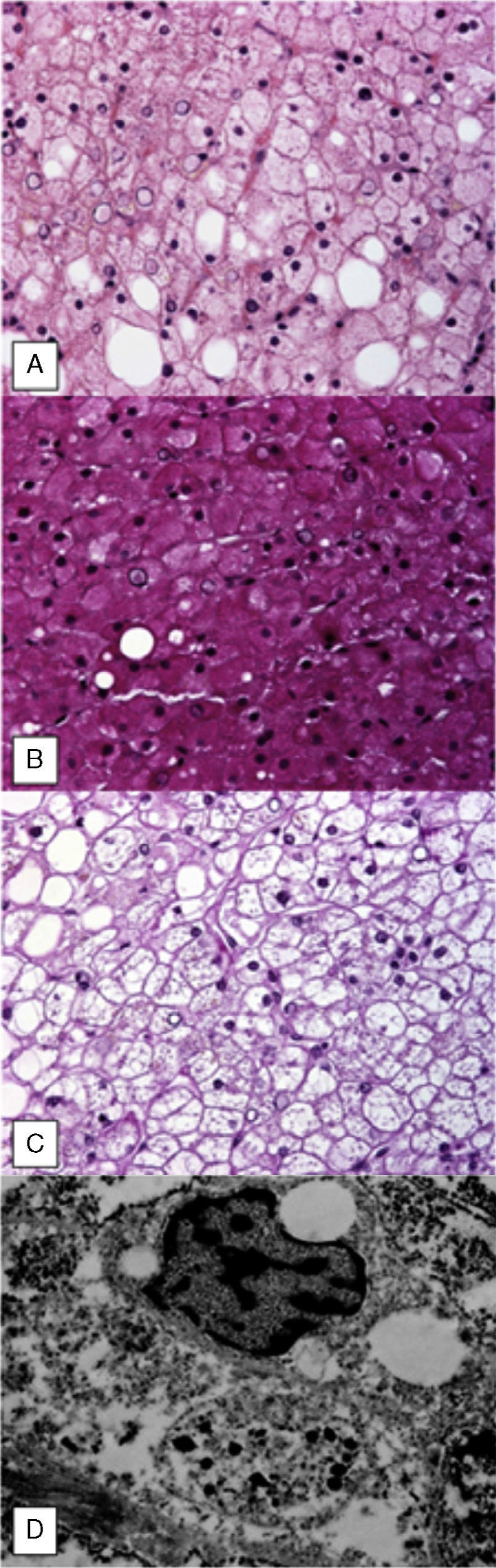
Ultrastructural evaluation confirmed the presence of nuclear and cytoplasmic glycogen deposits (fig. 2D).
Pierre Mauriac syndrome was described in 1930 and is characterized by retarded growth, a Cushingoid appearance, hepatomegaly, and hypertransaminasemia, mainly in chronically uncontrolled DM1 patients. Diagnosis requires a high index of clinical suspicion and is corroborated through liver biopsy; staining techniques such as PAS show the presence of glycogen, which disappears with the application of diastase, and steatosis is occasionally observed.
The patient's hepatomegaly and hypertransaminasemia were resolved after 4 months of optimum glycemic control, with HbA1c levels of 7%.
Financial disclosureNo financial support was received in relation to this article.
Conflict of interestThe authors declare that there is no conflict of interest.
Please cite this article as: Espinoza-Peralta D, Gutierrez-Llamas FJ, García-Juárez I. Síndrome de Pierre Mauriac y diabetes mellitus tipo 1 descontrolada. Revista de Gastroenterología de México. 2014;79:202–203.

