


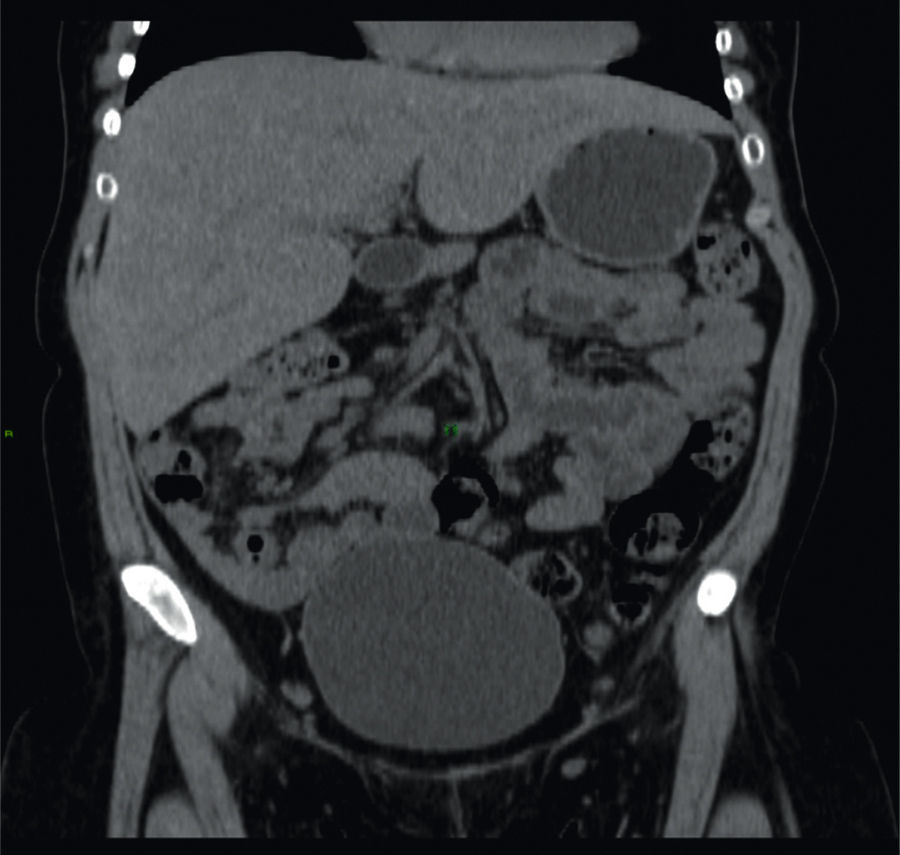
Mujer de 26 años de edad, con diagnóstico de diabetes mellitus tipo 1 (DM1) desde los 5 años. Descontrol crónico a pesar de múltiples esquemas de insulina con niveles de HbA1c 12%, conocida con retinopatía y neuropatía periférica. Cuadro clínico de un mes caracterizado por náuseas, vómitos y dolor abdominal. A la exploración presentó talla inferior a la esperada, sobrepeso, facies de aspecto cushingoide y hepatomegalia. Las pruebas de funcionamiento hepático solo mostraron aumento de transaminasas (ALT 158UI, AST 192UI) y FA 265UI. Se descartó etiología viral y autoinmune. Se realizó una TC simple de abdomen que reveló hepatomegalia de aspecto brillante (fig. 1).
En la biopsia hepática percutánea se evidenció una arquitectura normal con leves cambios por infiltración grasa (fig. 2A), en la tinción con ácido peryódico de Schiff (PAS) mostró abundantes depósitos de glucógeno (fig. 2B) que desaparecieron después de la digestión de diastasa (fig. 2C).
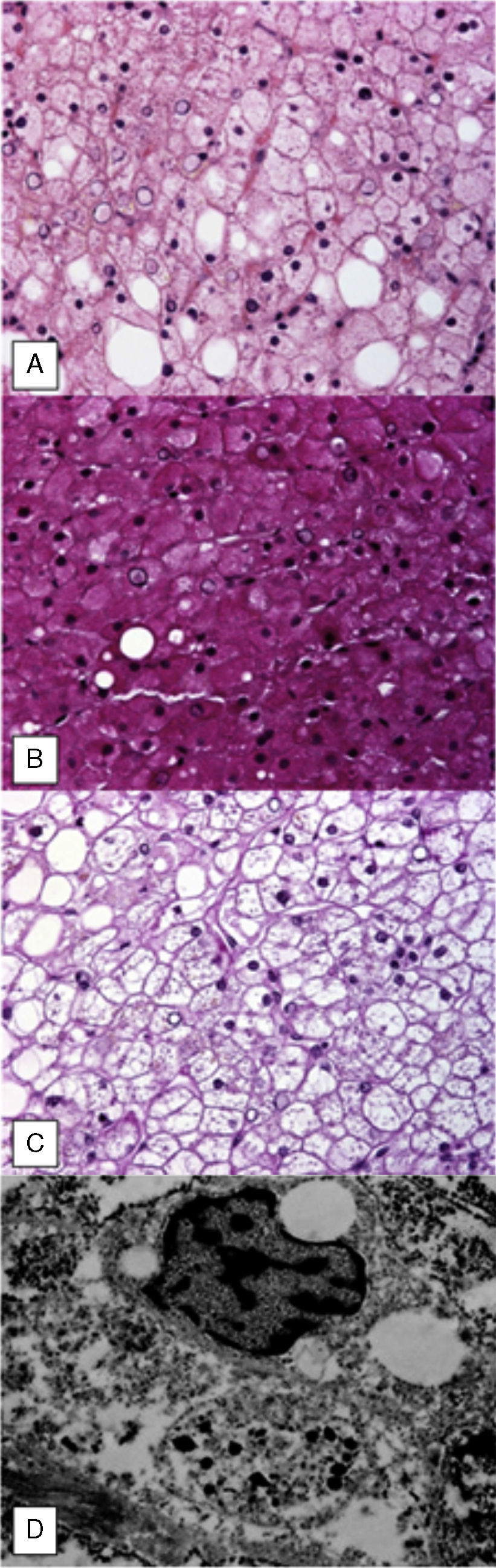
La valoración ultraestructural confirmó la presencia de depósito de glucógeno nuclear y citoplasmático (fig. 2D).
El síndrome de Pierre Mauriac fue descrito en 1930, caracterizado por retraso del crecimiento, apariencia cushingoide, hepatomegalia e hipertransaminasemia, principalmente en pacientes con DM1 y descontrol crónico. El diagnóstico requiere de alta sospecha clínica y corroborase en la biopsia hepática, con tinciones como PAS que muestra la presencia de glucógeno y la aplicación de diastasa hace que desaparezca y, ocasionalmente, puede observarse esteatosis.
Esta paciente presentó resolución de la hepatomegalia e hipertransaminasemia después de 4 meses de óptimo control glucémico HbA1c de 7%.
FinanciaciónNo se recibió patrocinio de ningún tipo para llevar a cabo este estudio/artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

