



Peutz-Jeghers syndrome (PJS) is an autosomal dominant inherited disorder characterized by hamartomatous polyps and mucocutaneous pigmentation, and its prevalence is 1 out of every 8,300 to 29,000 live births. The most frequently affected areas of the gastrointestinal tract are the small bowel, jejunum, and ileum (65-95%), and to a lesser degree, the colon (60%) and stomach (50%). The disease can give rise to complications such as intestinal lumen obstruction, bleeding, and a greater risk for intestinal and extra-intestinal neoplasias.1–3
A 30-year-old man was seen at the Instituto de Investigaciones Médico-Biológicas of the Universidad Veracruzana. The patient's family history included a father with intestinal polyps. He presented with a one-year progression of symptoms of abdominal pain, nausea, bloating, and constipation, accompanied with anorexia, adynamia, and weight loss. A relevant sign was the presence of hyperchromic spots on the upper lip and palate. Colonoscopy revealed numerous sessile polypoid lesions that varied in size from 0.3 to 2cm throughout the entire colon and in the terminal ileum. The histopathologic study reported pedunculated hamartomatous polyps with a stromal and epithelial histologic component and an arborescent proliferation of smooth muscle, resulting in the diagnosis of PJS.
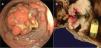
One year later, the patient returned complaining of persistent pain, now located at the left flank, abdominal bloating, important constipation, nausea and vomiting, and general malaise. A Pillcam (Given Imaging) capsule endoscopy was performed that showed numerous sessile polyps in the gastric chamber, duodenum, jejunum, and ileum. Passage to the colon was not possible due to the presence of a large polyp. In the following 7 days, the pain intensified and was accompanied with signs and symptoms of marked bowel obstruction, with no expulsion of the video capsule. The symptoms were corroborated through a plain abdominal film that showed the capsule located in the ileum. An antegrade double-balloon enteroscopy was then carried out, through which numerous bleeding ulcerated, eroded, and branched polyps were visualized, along with the video capsule that was lodged in the ileum. A large polyp did not allow the endoscope to pass, corroborating the bowel obstruction (fig. 1A). The maneuvers to extract the capsule were unsuccessful due to its impact on the stenosed site, and so the patient underwent surgical resection of a 77-cm segment of the terminal ileum with anastomosis of the ileum to the transverse colon (fig. 1B).
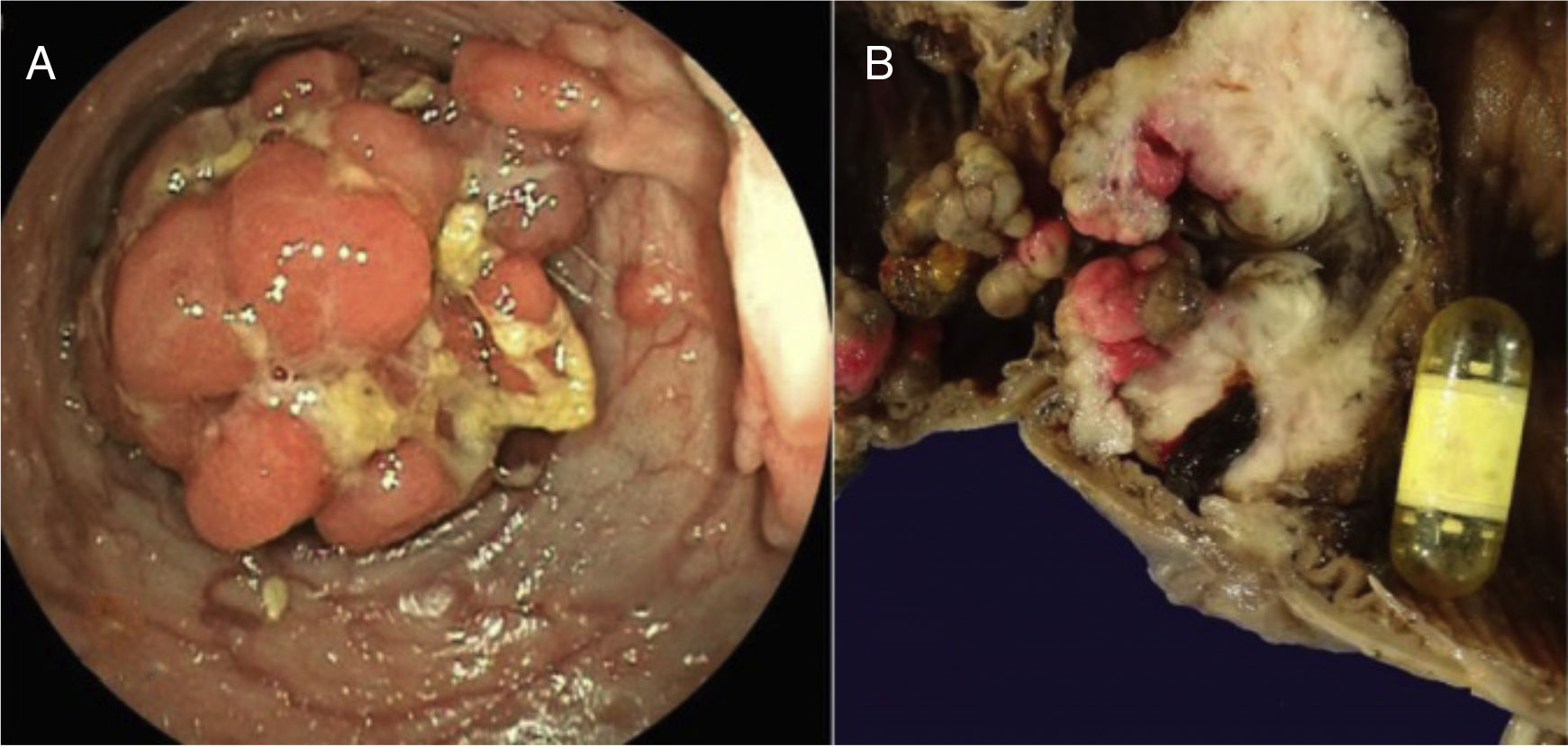
The histopathologic study of the specimen showed countless polyps measuring from 0.2 to 5cm in size. The largest one obstructed 90% of the lumen and the capsule was found in the proximal part of the lesion. Microscopic study revealed multiple ulcerated and bleeding polyps, and the stenosed lesion corresponded to a focal polyp with high-grade dysplasia and a stromal pseudo-invasive aspect (fig. 2A and B).
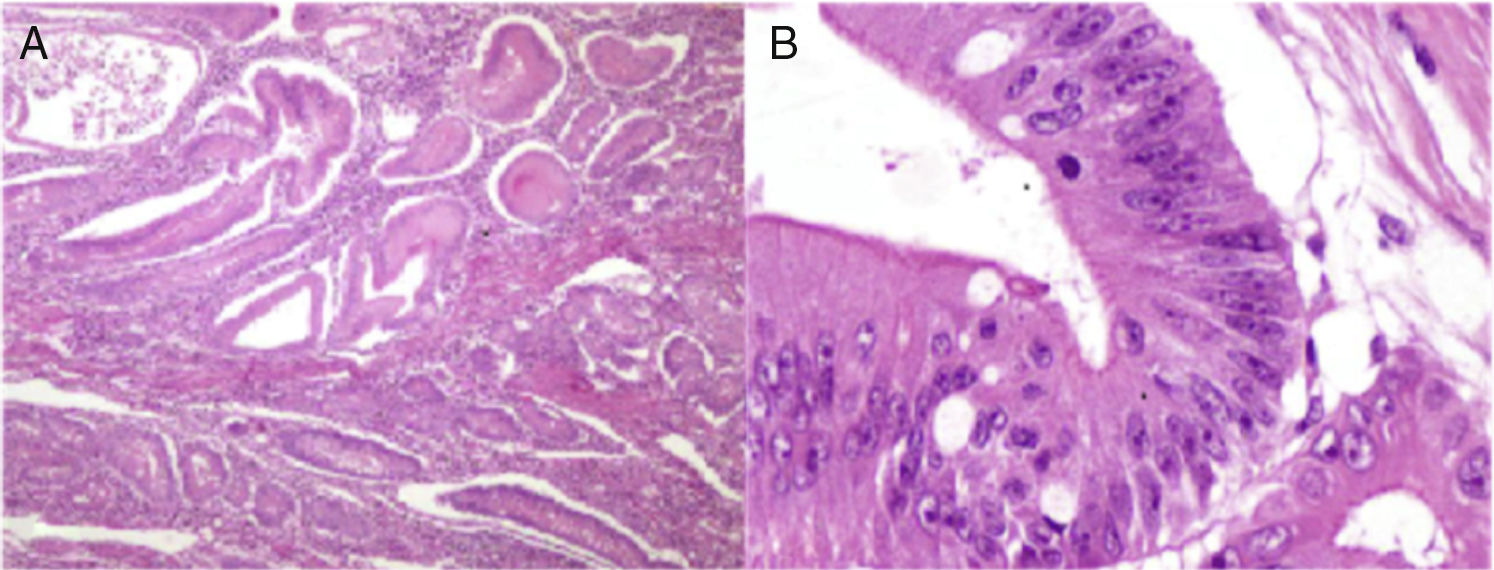
Histopathologic study of the lesion. A) Low-magnification photomicrograph showing the numerous Peutz-Jeghers polyps that have the characteristic thick bands of smooth muscle between the islets of epithelial tissue. B) High-magnification photomicrograph of the largest polyp with high-grade dysplasia.
After bowel resection, the patient presented with ischemic stroke in the right hemisphere associated with a patent foramen ovale, diagnosed through transesophageal ultrasound. It caused a reduction in the strength and sensitivity of the left limbs. Later progression was satisfactory, with considerable improvement of the patient's general state of health, as well as normal bowel transit. He underwent physical rehabilitation with good neuromuscular response.
In 1921, Peutz described the existing relation between mucocutaneous pigmentation and intestinal polyposis when studying 7 members of the same family. Years later, in 1949, Jeghers et al. published the autosomal dominant hereditary nature of the disease and the elevated risk for cancer in patients with the pathology. However, it was not until 1954 that Bruwer et al. first coined the name “Peutz-Jeghers syndrome”.4 Its incidence is low, and there is no predominance of sex or race. The average age at diagnosis is between 9 and 39 years, with a mean of 23 years.5 Seventy-five percent of the cases are the result of the dominant inheritance of the genetic mutation in the 19p 13.3 chromosome that encodes a serine-threonine kinase known as STK11/LKB1, and although its function has not been fully understood, it is thought to play a role in the development of cellular architecture causing tissue disorganization of variable penetration that gives rise to the formation of polyps at different locations, as well as to hyperchromic spots.6,7
Our case was of a young patient that met the criteria established by the World Health Organization (the presence of three Peutz-Jeghers polyps confirmed through histologic study, a family history of PJS, and mucocutaneous pigmentation)8 and presented with symptoms of bowel occlusion at the level of the terminal ileum due to polyposis, with video capsule impaction. Such impaction is rare and required resection of the affected bowel segment. The patient had a postoperative cerebrovascular accident due to a septic embolism through the foramen ovale that we do not believe was related to the gastrointestinal pathology.
Various authors of reports in the literature have stated that young patients with PJS have a high risk for developing cancer of the digestive tract (15-18 times higher than the general population), which is why our patient should undergo endoscopic follow-up to detect possible early malignization of the hamartomatous polyps.9,10
Financial disclosureOnly institutional funds were received in relation to this article.
Conflict of interestThe authors declare that there is no conflict of interest.
Please cite this article as: Roesch-Dietlen FB, Cano-Contreras AD, Meixueiro-Daza A, Remes-Troche JM, Grube-Pagola P. Obstrucción intestinal por cápsula endoscópica en un paciente con síndrome de Peutz-Jeghers. Revista de Gastroenterología de México. 2018;83:202–204.

