


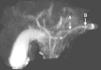


Chronic pancreatitis is a progressive inflammatory process of the pancreas that leads to fibrosis and exocrine and endocrine function loss with structural changes demonstrated by radiologic and histologic evidence.1,2
The clinical course of the disease is characterized by recurrent episodes of acute pancreatitis appearing at early ages, followed by a final stage with calcifications, pancreatic insufficiency, and diabetes mellitus in the majority of the patients.1
Chronic pancreatitis incidence due to any cause is from 3.5-10/100,000 inhabitants/year in Europe and the United States.3 Its cause cannot be defined in a significant percentage of cases (10-25%), resulting in the classification of idiopathic chronic pancreatitis. This is the classification in 40-60% of pediatric cases.4
Over the last few decades, in addition to the cystic fibrosis transmembrane conductance regulator (CFTR) gene, alterations have been identified in others, such as the cationic trypsinogen (PRSS1), anionic trypsinogen (PRSS2), serine protease inhibitor Kazal type 1 (SPINK1), and chymotrypsin C (CTRC) genes that play an important role in the pathophysiology of chronic pancreatitis, reducing the percentage of cases previously defined as idiopathic.4,5
We present herein the case of an 8-year-old boy that was admitted to the Hospital Pablo Tobón Uribe due to localized abdominal pain in the epigastrium, nausea, and vomiting. His nutritional status was adequate, epigastrium palpation was painful, but there were no other pathologic signs. He had a past history of an episode of acute pancreatitis at the age of 7 years and his family history included a brother with acute pancreatitis and no other relevant data. The patient's symptoms upon admittance were suggestive of uncomplicated acute pancreatitis and he had elevated pancreatic enzymes (amylase 1,359; lipase 3,098) (table 1). A nuclear magnetic resonance study of the pancreas revealed irregular dilation of the duct of Wirsung (fig. 1). Taking his past history and the biochemical and radiologic findings into account, the diagnostic approach for chronic pancreatitis was begun, ruling out other causes such as hypercalcemia and autoimmune pancreatitis; the quantification of immunoglobulin G subclass 4 was within normal parameters. Molecular studies were performed that included 60 mutations for CFTR, which were negative, and only the presence of the N34S mutation of the SPINK1 gene in exon 3 was reported. During the one year of follow-up at the institution, the patient once again presented with symptoms of uncomplicated acute pancreatitis and he has not presented with evidence of endocrine or exocrine insufficiency.
Paraclinical, imaging, and genetic studies performed.
| Paraclinical | Second episode | Third episode |
|---|---|---|
| Calcium | 9.4 mg/dl | 9.9 mg/dl |
| Amylases | 1,648-488-281-110-85 IU/l | 1,359-139 U/l |
| Amylase in urine | 343 | – |
| Lipases | 2,639-490-296-96-50 U/l | 3,098-221 IU/l |
| Albumin | 3.7 g/dl | – |
| Alkaline phosphatase | 169 IU/l | – |
| Gamma-glutamyltransferase | 14 U/l | 13 U/l |
| Cholesterol | 169 mg/dl | 184 mg/dl |
| Triglycerides | 182 mg/dl | 105 mg/dl |
| Glycemia | 86 mg/dl | 56-89-97 mg/dl |
| LDH | – | 200 mg/dl |
| BUN | 9 mg/dl | 7.7 mg/dl |
| CRP | 1.26 mg/dl | 0.11 mg/dl |
| ESR | 13 mm/h | – |
| Leukocytes | 3,800/m3 | 6,600/m3 |
| Neutrophils | 57% | 64% |
| Platelets | 188,000/m3 | 287,000/m3 |
| Iontophoresis | Negative | |
| Stool analysis | No fats | |
| CFTR (60 mutations) | Negative | |
| PRSS1 (exon 2 and 3) | Negative | |
| SPINK1 (exon 3) | C. 101A>G/p. Asn34Ser. | |
| Immunoglobulin G subclass 4 | 12 mg/dl | |
| Contrast and non-contrast magnetic resonance cholangiography | Pancreas with beading of the duct of Wirsung, normal parenchyma. No congenital biliopancreatic abnormalities or lithiasis | |
| Complete abdominal ultrasound | Normal |
BUN: blood urea nitrogen; CFTR: cystic fibrosis transmembrane conductance regulator; CRP: C-reactive protein; ESR: erythrocyte sedimentation rate; LDH: lactate dehydrogenase; PRSS1: cationic trypsinogen; SPINK1: serine protease inhibitor Kazal type 1.
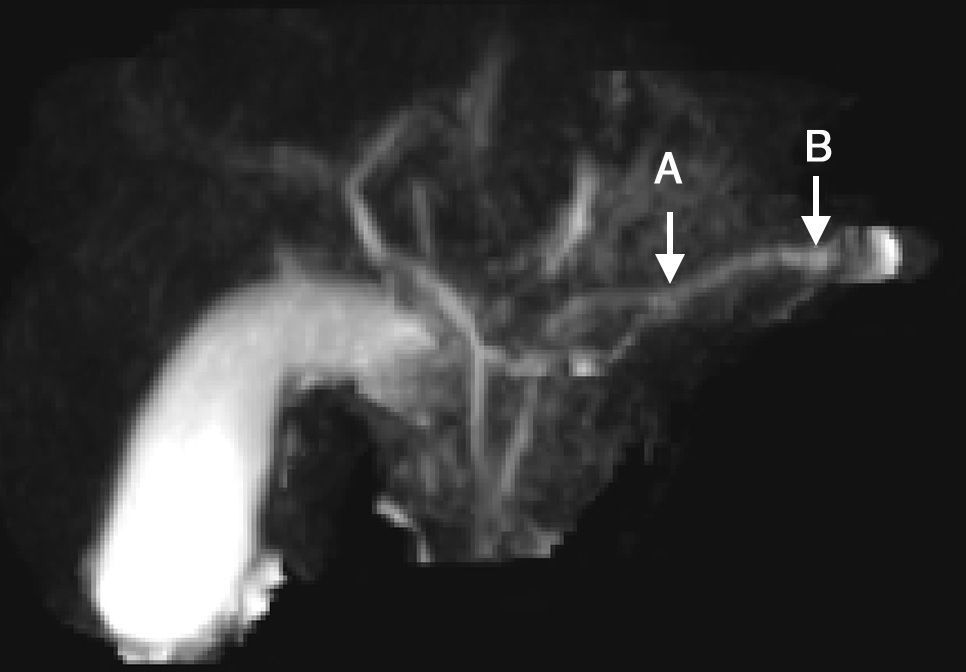
This patient has had 3 episodes of acute pancreatitis with asymptomatic intercritical periods, irregular dilation of the duct of Wirsung revealed by imaging studies, and identification of the N34S mutation of the SPINK1 gene. This mutated gene has been associated with recurrent acute pancreatitis, as well as with hereditary chronic, alcoholic, tropical calcifying, and idiopathic pancreatitis.3 It has also been associated with higher rates of pancreatic lithiasis, pseudocysts, and calcifications.4 Likewise, it has been seen in a small percentage of asymptomatic controls and in other pancreatic pathologies, suggesting that these mutations are modifying factors, rather than causes, of the disease. In our case, the SPINK1 alteration was the only cause that could be attributed, with the limitation that other genes, such as the CTRC, and other exons of the PRSS1, were not studied.
The most frequent clinical presentation is early age onset, around 10 years of age, with intermittent symptoms of nausea and vomiting, in which the abdominal pain may or may not be the main symptom, and a slow progression toward pancreatic insufficiency and endocrine function compromise that can occur at more advanced ages, with radiologic evidence of structural changes (irregular dilation of the pancreatic ducts, atrophy, glandular calcification, and peripancreatic fat alterations).2,3,5,6
The pathophysiologic process of this entity is still under study. Current findings point to a process of autodigestion due to the premature activation of trypsinogen secondary to mutations in the genes that encode proteins that are in charge of keeping it inactive while it is in the pancreas (fig. 2). PRSS1 alterations have been described as the main mutations associated with chronic pancreatitis, but in the last few years, association with other genes, such as the PRSS2, SPINK1, and CTRC, has been described.7–9
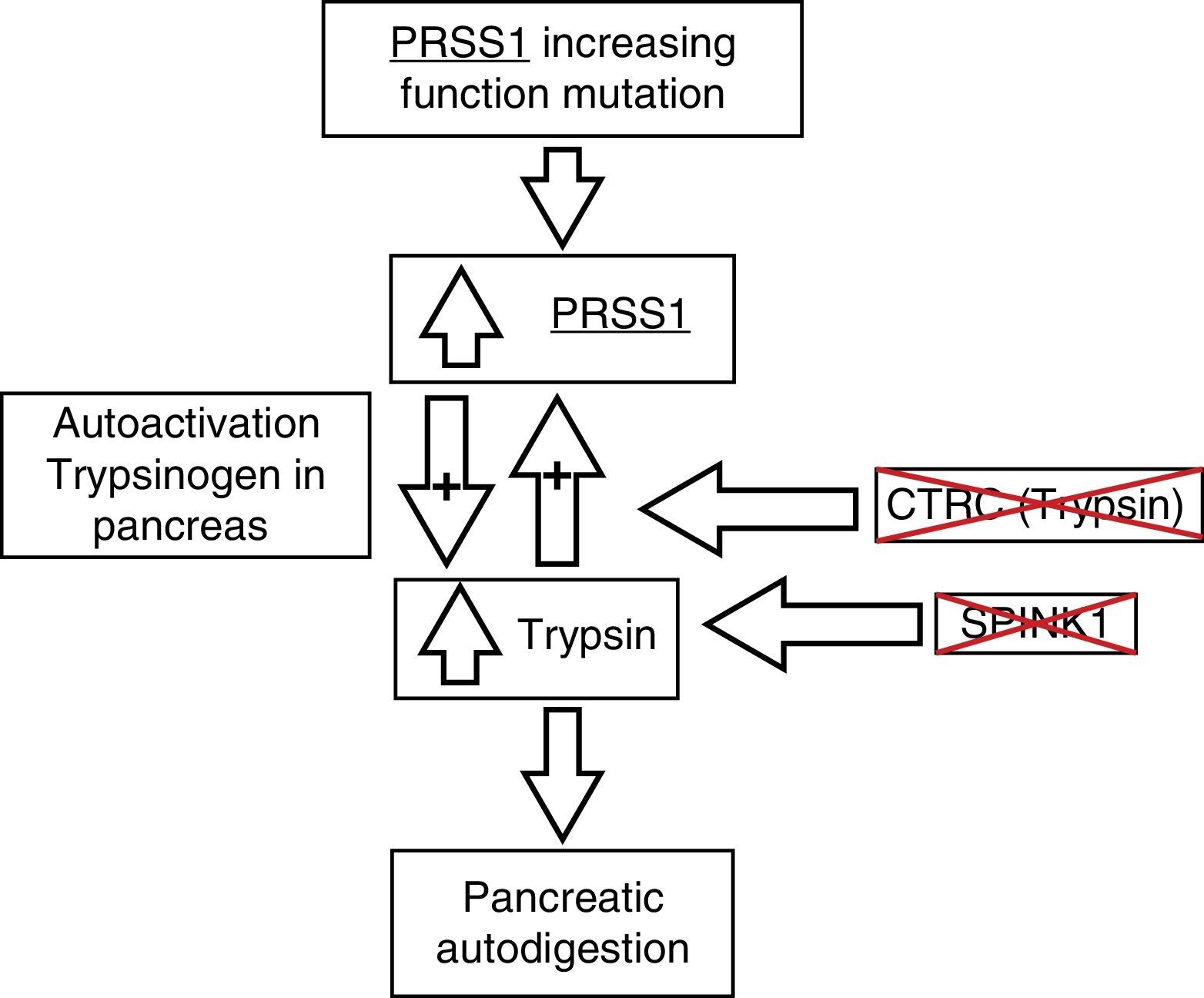
Pathophysiology of chronic pancreatitis and its genetic bases. CTRC: chymotrypsin C; PRSS1: cationic trypsinogen; PRSS2: anionic trypsinogen; SPINK1: serine protease inhibitor Kazal type 1. Adapted from Derikx et al.7
SPINK1 acts as the first line of defense against trypsinogen and is capable of inhibiting around 20% of the total activity of trypsin within the pancreas. The most commonly implicated mutation of this gene is the N34S mutation, according to recent data from genetic studies of patients with chronic hereditary and idiopathic pancreatitis.3,5,6,10
The prevalence of these mutations varies in accordance with the geographic area. Latin American case series have been published that report these gene mutations in the pediatric population with recurrent pancreatitis; the most frequent is the PRSS1 gene mutation, whereas the SPINK1-NS34 mutation has been found in very few cases.11–13
The prevalence of hereditary chronic pancreatitis is low in the pediatric population. Its diagnosis is limited due to a low rate of suspicion and the difficulty in our environment of identifying the specific gene mutations,5 giving relevance to the publication of clinical cases such as this one. Descriptions of the typical characteristics of this pathology and a complete diagnostic approach enable possible complications to be anticipated, environmental risk factors to be avoided, and family genetic counseling to be provided.
Financial disclosureNo financial support was received in relation to this article.
Conflict of interestThe authors declare that there is no conflict of interest.
Please cite this article as: Ruiz-Navas P, Contreras-Ramirez M, Montero-Carvajalino AE, Castaño-Jaramillo LM, Orozco-Forero JP. Mutación de SPINK1 en paciente pediátrico con pancreatitis crónica. Reporte de un caso. Revista de Gastroenterología de México. 2015;80:223–225.

