
Understanding of the pathophysiology of hepatic encephalopathy has conditioned new treatment options. Ammonia detoxification in hepatic encephalopathy is regulated by two enzymes: glutaminase or glutamine synthetase. The first produces ammonia and the second detoxifies the ammonia, which is why treatments are aimed at glutaminase inhibition or glutamine synthetase activation. At present, we know that both enzymes are found not only in the liver, but also in the muscle, intestine, kidney, and brain. Therefore, current treatments can be directed at each enzyme at different sites. Awareness of those potential treatment sites makes different options of approach possible in the patient with hepatic encephalopathy, and each approach should be personalized.
El entendimiento de la fisiopatología de la encefalopatía hepática ha condicionado nuevas opciones de tratamiento. La detoxificación del amonio es regulada por 2 enzimas en la encefalopatía hepática: la glutaminasa o la glutamina sintetasa. La primera genera amonio y la segunda detoxifica el amonio, por lo que los tratamientos son encaminados a la inhibición de la glutaminasa o a la activación de la glutamina sintetasa. Actualmente sabemos que ambas enzimas se encuentran no solo en el hígado, sino también en el músculo, intestino, riñón y cerebro, por lo que los tratamientos actuales pueden ser dirigidos a cada enzima en sitios separados. Entendiendo estos sitios potenciales de tratamiento, las opciones para el abordaje del paciente con encefalopatía hepática son diversas y deben ser personalizadas.
Hepatic encephalopathy (HE) presents in 30-60% of patients with cirrhosis of the liver. It is a clinical expression of a spectrum of potentially reversible neuropsychiatric abnormalities secondary to the accumulation of neurotoxic substances in brain tissue that is proportional to the synthetic function and functional reserve of the liver. Therefore, it is important to know and understand its components, pathophysiology, and therapeutic targets, so that the risk for complications (hospital-acquired pneumonia, inadequate secretion management, sarcopenia, and malnutrition, among others) is reduced and the functional class of the patients can be improved.
HE is classified according to 4 factors: the underlying disease, the severity of the manifestations, the time course, and the precipitating factors. There are 3 types of underlying disease, depending on the clinical context in which it occurs: type A presents in the context of acute liver failure, type B in the context of porto-systemic shunting with no intrinsic liver disease, and type C in the context of cirrhosis with portal hypertension. Disease manifestations are classified as covert, grade I, grade II, grade III, and grade IV, according to the patient's clinical characteristics (behavioral changes, confusion, bradypsychia, sleep-wake cycle alterations, incoherent speech, lethargy, stupor, and coma) and neuropsychologic or psychometric alterations. The time course of the disease can be episodic (2 or more episodes of HE within the span of 6 months or less) or persistent (a pattern of altered behavior that is always present, with no periods of normalcy). Finally, HE episodes are described as nonprecipitated or precipitated, in relation to whether a triggering factor is found or not.1
Pathophysiology of encephalopathyHistorically, the presence of elevated toxin levels in the cerebral parenchyma have been involved in the pathophysiology of HE, with ammonia (NH3) as the main agent. Current evidence supports the idea that ammonia is only one component of the numerous pathophysiologic factors that contribute to the appearance of HE.2–9
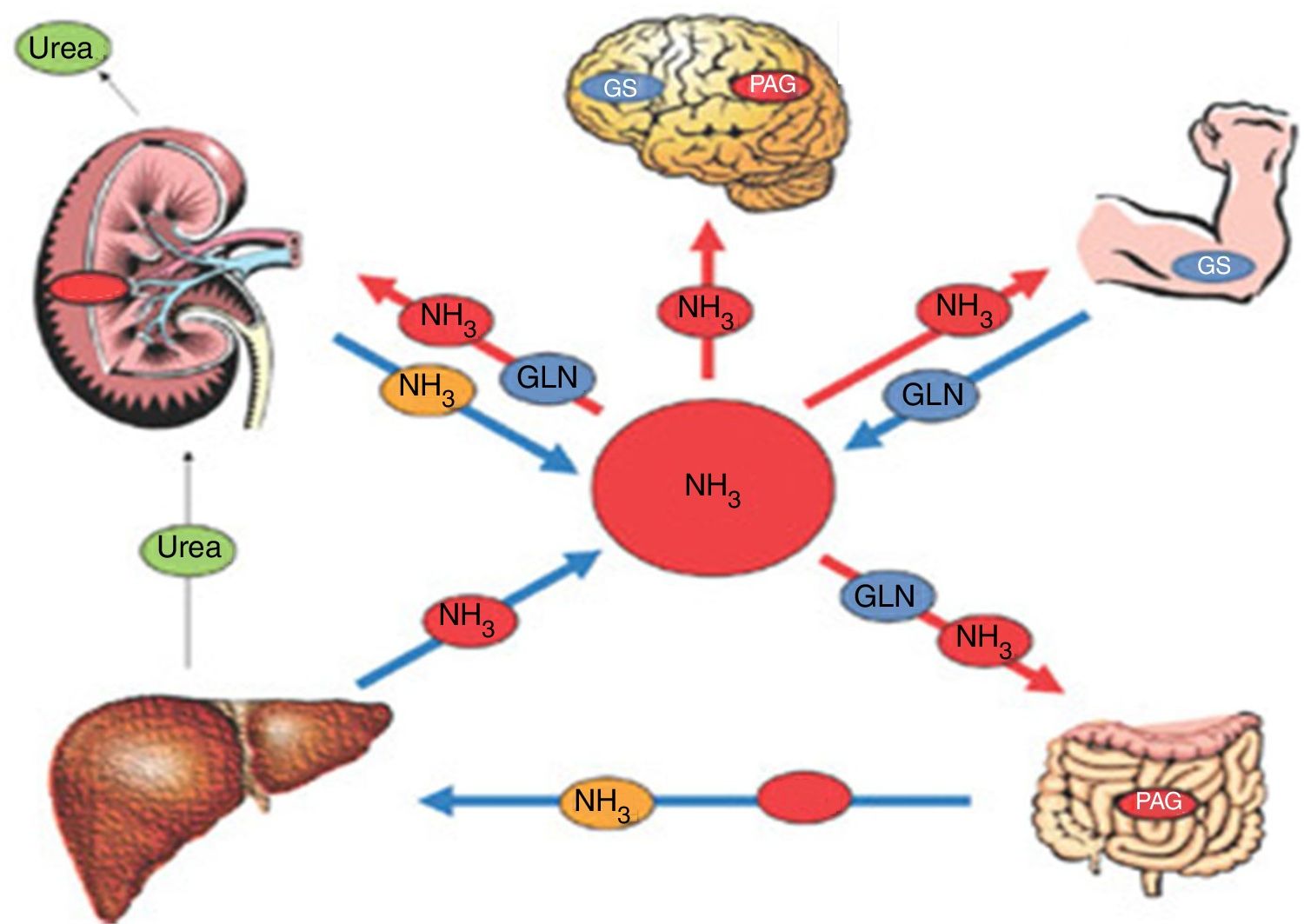
The pathophysiologic approach should include the integrity of the natural detoxification sites of the liver, intestine, muscle, kidney, and astrocyte, as well as the study of the collateral circulation (fig. 1).

Pathophysiology of HE. In patients with cirrhosis, the concentration of ammonia can increase significantly. That is due to less detoxification of ammonia in the liver and to some other metabolic alteration in other organs, such as the muscle, kidney, or intestine, impeding the adequate elimination of ammonia.
GLN: glutamine; GS: glutamine synthetase; NH3: ammonia; PAG: phosphate-activated glutaminase.
Source: adapted from Rose CF. Ammonia-lowering strategies for the treatment of hepatic encephalopathy. Clin Pharmacol Ther. 2012;92:321-331.
Classifying the severity of liver dysfunction, based on the Child-Pugh scale, in patients with cirrhosis, enables synthetic function to be calculated and the complications due to hepatopathy to be evaluated. There is concern about the accumulation of endogenous neurotoxins (NH3) in patients with a Child B or C classification or with decompensated cirrhosis. Even though patients with a Child A classification have a much lower percentage of clinical encephalopathy, we should intentionally search for minimal HE through the adequate tests because treatment at that phase can impede progression to obvious clinical forms.10
Ammonia in the liver is eliminated largely through the urea cycle. Urea is the main product of protein degradation produced by the liver through that metabolic pathway. L-ornithine L-aspartate (LOLA) functions at that site as a substrate of the urea cycle and thus intervenes in the lowering of ammonia levels through an increase in the flow of glutamine synthetase (GS) and the urea cycle enzymes. Several researchers have conducted meta-analyses to study the effectiveness of LOLA in cirrhotic patients with HE, subdivided into overt HE and minimal HE. The studies concluded that LOLA has beneficial effects on both groups, with respect to improvement in mental status, HE grade, and serum ammonia levels.11,12
Zinc (Zn) is an important cofactor in the enzyme reactions carried out in both the liver and muscle. At the molecular level, a lack of Zn is thought to reduce the action of ornithine transcarbamylase, an enzyme of the urea cycle, and GS, an essential enzyme in nitrogen metabolism. Those 2 enzymes are in charge of the elimination of ammonia in the liver and muscle, respectively, and Zn supplementation is accompanied by an increase in their enzyme activity. Some studies have shown important Zn depletion in patients with cirrhosis13 and 95% of cirrhotic patients with a MELD score equal to or above 15 points are estimated to have Zn deficiency,14 which correlates with the greater incidence of encephalopathy in those patients. In their study, Bresci et al. reported improvement in the psychomotor tests in the group of patients with cirrhosis that received Zn supplementation, compared with the group that did not receive it, but unfortunately the difference was not statistically significant.15 In another review article on Zn deficiency, Zn supplementation was suggested for consideration as a therapy in patients that did not respond to treatments based on low-protein diets and the administration of lactulose.16
The benefit of Zn was demonstrated later by the Takuma group through a clinical trial on treatment-refractory cirrhotic patients with grade I and grade II HE. In that 2-arm study, the patients were divided into the group receiving Zn supplementation and standard therapy (branched-chain amino acids and lactulose) and the group receiving only standard therapy. The patients were followed for a period of 6 months to determine the effects of HE on quality of life. The patients that received the supplementation showed significant improvement with respect to the physical component scale, the grade of encephalopathy, and blood ammonia levels. The grade of encephalopathy improved in approximately 54% of the patients and HE grade 0 was achieved in 41%. It should be mentioned that the masking technique was a limitation of that study because it was not a blind analysis and the Zn supplement contained L-carnitine, which can act as an antioxidant.17
The following aspects are among the disadvantages of Zn supplementation: the action of ciprofloxacin is reduced, the effective dose has not been found due to the lack of studies, and its prolonged use produces copper deficiency and dyspepsia.
IntestineThe overproduction of ammonia in the intestine plays a key role in the development of HE. The scientific bases were posited at the middle of the past century upon the realization that HE was related to the absorption of nitrogenous substances coming from the intestine. Around the same time, methionine was identified as a toxic agent and the abundance of coliform bacteria that resided in the small bowel in patients with cirrhosis was described. Bajaj et al. have since identified different changes that take place at the level of the gut microbiota in patients with cirrhosis and realized that there is greater dysbiosis when those changes occur at a more decompensated disease phase. Thus the cirrhosis dysbiosis ratio was established, in which a lower ratio is associated with greater morbidity and mortality.18,19 At present, only Chen and his research group have been able to reproduce the fecal microbiome of the cirrhotic patient.20 That is important because each population is different, and thus the predominant microbiota should be the focus of potential therapeutic targets.
The approach based on poorly absorbed antibiotics, such as rifaximin or neomycin, has historically been used to reduce intestinal ammonia produced by the urease-producing bacteria. There is little evidence on the efficacy of neomycin, but rifaximin is one of the most widely studied therapeutic options. Rifaximin in the treatment of patients with HE has been evaluated in numerous studies, confirming its efficacy and safety at doses of 1,200 or 2,400mg/day.21–27 Its usefulness in secondary prophylaxis has also been corroborated.26 Other analyses have compared rifaximin with nonabsorbable disaccharides and both have been shown to be efficacious as treatment for HE, reducing the blood ammonia levels and improving symptoms. Nevertheless, rifaximin has shown earlier improvement and fewer side effects.23,25,27 Likewise, the use of oral vancomycin was reported to reduce the diversity of the gut microbiota.28 In a Japanese study on 12 patients with lactulose-resistant HE, vancomycin was added to the management and the HE grade improved.29 The results of that small blind study suggest that vancomycin could be an option in patients that do not respond to treatment with lactulose, but that should be cautiously interpreted, given the design of that study and the fact that there is scant evidence on the use of said drug.
Nonabsorbable disaccharides, such as lactulose (β-galactosidofructose) and lactitol (β-galactosidosorbitol) are able to reduce both the intestinal production and absorption of NH3 through acidification in the colon, which is a laxative effect, and the movement of NH3 from the portal circulation to the colon. They also interfere with glutamine uptake by the intestinal mucosa and the subsequent reduction of NH3 synthesis and absorption. The dose of lactulose (15 to 30ml administered 2 to 4 times a day) is titrated so that the patient has 3 to 5 bowel movements daily. It can be administered rectally in patients that cannot take oral medications. Those two nonabsorbable disaccharides are considered first-line therapy for the treatment of HE and there is symptom improvement in about 70% of cases. Numerous studies have evaluated lactulose in the treatment of minimal HE, overt HE, and as a secondary prophylactic measure in patients with cirrhosis, confirming its efficacy and safety.30–33 Clinical trials have shown that lactitol is better-tolerated and is just as efficacious as lactulose for the treatment of HE.34,35 No significant difference has been found with respect to improving HE grade in the comparison of nonabsorbable antibiotics (rifaximin) versus nonabsorbable disaccharides (lactitol and/or lactulose).23,25,27
Probiotics are mixtures of beneficial bacteria that are believed to aid in the treatment of HE by modulating the microbiome. In relation to that possible favorable effect, the studies that focus on utilizing probiotics to improve the gut microbiota in patients with cirrhosis offer an interesting alternative for modulating the bacterial flora of those patients. There is no evidence on the use of probiotics in the acute treatment of overt HE. In the principal studies on the use of probiotics in overt HE, they were employed as secondary prophylaxis and were effective for preventing HE in patients with cirrhosis.36,37
MuscleSkeletal muscle has been described as the second buffer in NH3 detoxification. The myocytes provide a site for metabolizing NH3, incorporating it into glutamine through GS, even though the activity of that enzyme in the muscle is low. In a certain way, NH3 clearance and glutamine production can make up for the lack of metabolism by the liver but that cannot be the solution to hyperammonemia because of NH3 overproduction at other sites.
Malnutrition in cirrhotic patients and their loss of muscle mass perpetuate and worsen the appearance of HE. In addition, catabolism per se produces excess glutamine in the circulation, which leads to greater renal and intestinal production of ammonia. Nutritional management in patients with cirrhosis is an essential part of the preventive management of HE. The current recommendation is for patients with HE to maintain the same diet as other patients with cirrhosis, because there is no evidence that dietary protein restriction prevents episodes of HE.38,39 Despite the effort to maintain their nutrition, certain patients with cirrhosis present with sarcopenia and there is evidence suggesting that said loss of skeletal muscle mass is associated with the risk for developing minimal HE or overt HE.40,41 In theory, HE prevention could be helped by attempting to improve nutritional status and muscle mass through a protein-rich diet and a high-protein snack before going to bed.38,42 In addition, exercise and branched-chain amino acids have been studied as therapeutic options for treating sarcopenia in patients with cirrhosis and the results have been satisfactory.43 Branched-chain amino acids were also analyzed in a Cochrane review as a therapeutic option for HE, and they were shown to significantly increase the number of patients that had HE improvement.44
Increased expression of myostatin or growth differentiation factor 8 (GDF-8) belonging to the transforming growth factor-β (TGF-β) family of proteins was shown in the muscle mass of cirrhotic patients. That protein is known for its inhibitory properties in muscle production and growth due to its historic relation to the activin type IIB receptor (ActRIIB), achieving said repression by increasing the Smad 2/3-mediated transcription. It has been shown to be secondary to intramuscular NH3 accumulation, resulting in activation pathways that contribute to sarcopenia.45
In that line of thought, it is easy to imagine that the inhibition of ActRIIB or the minimization of GDF-8 expression could be new treatment alternatives. In their study, MacKenzie et al. showed the benefit of inhibiting GDF-8 expression through resistance exercise, which correlated with an increase in muscle mass.46
In 2012, Duarte-Rojo et al. studied the changes in GS messenger ribonucleic acid of mononuclear cells in peripheral blood after exercise in healthy volunteers. They found that GS was indeed more highly expressed under conditions of exercise and distributed in the cytoplasm of peripheral blood mononuclear cells. Those results are encouraging for the treatment of HE due to their implications in NH3 metabolism.47
KidneyThe kidneys also play an essential role in ammonia homeostasis. Under normal conditions, renal cells produce NH3 ions that can take 2 routes: they can be excreted in urine or reach the blood circulation through the renal vein. In healthy persons, the latter mechanism represents the largest part of the circulating concentration of NH3, and its excretion into the renal vein can be modified according to the acid/base status, potassium levels, and kidney function. Even though glutamine is the main tributary amino acid in NH3 production, glutamate, glycine, and proline are the amino acids that are equally involved in that task. In patients with cirrhosis, glutamine has been shown to be the alternate route of greater action for detoxifying ammonia, and under conditions of stress, can remove up to 80% of NH3.48 Finally, as mentioned above, diet and exercise play an essential role in those patients.
Glycerol phenylbutyrate (GPB) is a medication approved by the Food and Drug Administration in 2013 for the treatment of patients with urea cycle disorders or innate metabolism errors that manifest as hyperammonemia, whose effect was emulated in the cirrhotic population.49 GPB is hydrolyzed by pancreatic lipases for producing glycerol and 3 molecules of phenylbutyric acid. The latter suffer β-oxidation in the liver to produce phenylacetic acid that is conjugated with glutamine in the liver and kidney, forming phenylacetyl glutamine that is then excreted in urine. That drug acts by providing an alternative route for the elimination of NH3 and the elimination of residual nitrogen in the form of urinary phenylacetyl glutamine. Consequently, an attempt was made to emulate that effect in patients with cirrhosis and HE in a pilot study, and GPB was shown to be capable of reducing NH3 levels in those patients.50 A later study by Rockey et al. was conducted on the usefulness of GPB in patients with cirrhosis that had presented with 2 or more HE events in the last 6 months. They demonstrated that GPB reduced the number of patients that experienced a new HE event, the total number of HE events, and serum ammonia levels.51 Those results are encouraging and suggest that GPB could be a potential therapeutic option for HE in patients with cirrhosis.
Acetyl-L-carnitine (ALC) is a short-chain ester derived from carnitine that is produced endogenously in the mitochondria and peroxisomes. It is involved in the transport of acetylic particles through the membranes of those organelles. The administration of that ester in animal models reduced NH3 toxicity through the activation of urea cycle enzymes, the interaction with glutamate receptors, and the reduction of free radicals.52–54 Studies have evaluated the use of ALC in patients with cirrhosis and occult and/or overt HE, showing that ALC produced improvement in quality of life, anxiety, depression, and cognitive functions.55–58 Those studies demonstrated that the administration of ALC reduced NH3 levels and had a protective effect against ammonia toxicity and glutamate neurotoxicity. Finally, more studies are needed to be able to recommend its use, but that intervention could be considered a future therapeutic target.
BrainEvidence of neuro-aggression includes the activation of the microglia, as well as the in situ synthesis of proinflammatory cytokines, tumor necrosis factor (TNF), interleukin 1 β (1L-1 β), and interleukin 6 (IL-6).59 The signaling mechanisms in liver failure include: the direct effects of systemic proinflammatory molecules, monocyte recruitment after microglial activation, accumulation of cerebral NH3, lactate, manganese, mercaptans, some fatty acids and tryptophan derivatives as neurotoxic substances, and the alteration in the permeability of the blood-brain barrier. As is well-known, both proinflammatory molecules and ammonia work together, causing cerebral edema.
Current anti-ammonemic strategies have had very little positive impact on the quality of life of cirrhotic patients, and so new therapeutic targets are being studied. Evidence points to both central and peripheral proinflammatory mechanisms working alone or together with neurotoxic molecules, such as NH3.59,60 In turn, hyponatremia, sepsis, gastrointestinal bleeding, and kidney failure precipitate the exogenous production of TNF, which exacerbates HE presentation.
Microglia are defense cells of the central nervous system (CNS) that have a great ability to recognize a wide range of homeostatic changes, from endothelial or tissular damage to changes in cellular energy capacity. Once they are activated, they produce a large cascade of cytokines and chemokines, with proinflammatory and inflammatory properties, depending on the situation. That was demonstrated in the autopsies of 8 patients with cirrhosis that died from liver failure.61 The extension of microglial activation and elevated mRNA levels of proinflammatory cytokines were shown to be predictors of HE, as well as of cerebral edema. Microglial recruitment was related to elevated levels of TNF and the consequent formation of CC-motif chemokine ligand 2 (CCL2) that forms part of the so-called chemokines.62 Those findings have suggested that there is a new immune-to-brain communication pathway, resulting in altered excitability and neurologic complications in patients with cholestatic liver disease. CCL2 inhibition has been shown to notably reduce progression to encephalopathy in animal models. Microglial activation has also been shown to be predominant in the frontal lobe. Cerebral GS is produced mainly by the microglia and is one of the first cerebral detoxification mechanisms. Therefore, the anaplerotic flow between the glutamate-glutamine cycle and the citric acid cycle, mediated by the pyruvate carboxylase enzyme, has been proposed to have detoxification capacities.63
Comparing the susceptibility to morphologic and functional changes of the astroglial and microglial cells under hyperammonemic conditions, astrocytes are more susceptible.64 The capacity of ammonia to free proinflammatory substances on the part of the microglia has also been studied in that context. Up to now, the results have not shown a direct relation, and more in vivo and ex vivo studies are needed in directed regions of the brain. Great synergy between NH3 and proinflammatory cytokines, especially TNF, has been described in studies conducted on patients with cirrhosis.59
As mentioned before, lactate also plays an important role in neuro-inflammation. There is solid evidence that lactate levels correlate with elevated NH3 concentrations in the brain in patients with terminal liver disease. Likewise, lactate is related to microglial activation, clinical symptom severity, and changes in the electroencephalogram, because there is a connection between the citric acid cycle and the urea cycle. Thus, the accumulation of lactate has the capacity to inhibit NH3 production by deactivating alpha-ketoglutarate dehydrogenase, which can activate the regulatory enzyme of the glycolytic pathway, phosphofructokinase 1. As a consequence, that leads to more lactate production and perpetuates the damage.65
One of the more striking findings in the post mortem brains of cirrhotic patients is the large quantity of manganese accumulated in the basal ganglia. That accumulation occurs thanks to 2 pathways: the first is the poor or null elimination of that metal by the biliary tract, which causes an elevated serum concentration, and the second is portal hypertension and the creation of collateral circulation, favored by the disruption of the blood-brain barrier, as the pathway of propagation to the brain. Manganese, together with oxidative/nitrosative stress, has been identified as the main agent of selective death of the cerebral dopaminergic cells, which is consistent with the relation found between cirrhosis and parkinsonism.66–68
Without a doubt, the final neuroprotection barrier is the blood-brain barrier, in the context of patients with cirrhosis. Recent studies have confirmed that, when exposed to a proinflammatory environment (TNF and IL-1 β), that barrier tends to lose its contention containment capacity, favoring the passage of toxins coming from the rest of the economy, thus becoming another harmful pathway between the liver and brain. The cause of that permeability is still being studied. Some theories suggest that cell adhesion proteins could play an important role in the origin of that phenomenon.69
Pathophysiologic points not directly related to ammoniaCerebral vasoreactivityCerebral hemodynamics is involved in the pathophysiology of HE through the direct damage to the vascular endothelium, endogenously producing higher levels of TNF and IL-1 β, as shown in the study by Macías-Rodríguez et al. Through transcranial Doppler ultrasound measurements of flow velocity, they demonstrated altered cerebral hemodynamics in patients with cirrhosis.70 Those findings correlated with severity measurements determined by the MELD scale and the MELD-Na variable. The study conclusion stated that both structural vascular damage and loss of cerebral blood flow autoregulation could be part of the pathophysiology of HE.
HyponatremiaLow sodium levels are common in the cirrhotic population due to the activation of the antidiuretic hormone, produced by a diminution in the effective arterial volume related to splanchnic vasodilation. When that state becomes chronic, it leads to an intracellular depletion of organic osmolytes, such as myo-inositol, that plays an important role in intracellular hydroregulation. The osmolytes in the astrocytes provide cellular defense against edema and can be rapidly accumulated or depleted, according to osmotic sensors. One of the theories about astrocytic swelling is the presence of chronic hyponatremia that causes the depletion of those osmolytes. Hence, with the permeability in the blood-brain barrier and the vulnerability to the hyperammonemic and inflammatory environment, low-grade edema and oxidative/nitrosative stress are produced, with astrocytic dysfunction.71
New therapeutic targetsManganese-chelating agentsBased on the evidence suggesting the involvement of manganese in the death of dopaminergic cells and its accumulation reported in the brain tissue of cirrhotic patients, it is not illogical to think that manganese chelators could aid in lowering the rates of parkinsonism associated with cirrhosis and in reducing the expression of oxidative stress radicals produced by their interaction with the microglia. Ethylenediaminetetraacetic acid (EDTA) and p aminosalicylic acid (PAS) have been studied with respect to that, but more analyses need to be conducted.72
Urea transporter proteinsAs we know, urea is a water-soluble molecule whose motility has classically been described as passive diffusion. Currently, the cloning of the specific proteins that facilitate urea transport in certain tissues has had a radical effect on our understanding of the physiologic importance of that molecule. The genes responsible for encoding those proteins have been found (SLC14A1 and SLC14A2) and they form part of the same chromosome (18q12.q21.1). In the context of HE, the novelty of that discovery is that their possible blockage could stop the increased production of intestinal NH3 (UT-B urea transporter) and the stimulation of the vasopressin-regulated UT-A 1 and 3 isoforms as urea inducers in the kidney medulla.73
TaurineOne of the interesting points in the analysis of HE pathophysiology is the low concentration of taurine. At the level of the brain, that amino acid acts as a powerful antioxidant and as a nitric oxide synthetase regulator. In addition, it possesses antimicrobial properties, increasing the phagocytic ability of the neutrophils and the protection of the endothelium.74
FollistatinFollistatin is a glycoprotein and member of the TGF-β family that antagonizes many members of that family, including activin A, growth differentiation factor 11 (GDF-11), and GDF-8. Administration of that molecule produced an increase in body weight dependent on muscle mass in rats. It has also improved aggression and muscle atrophy, modulating the early response of the inflammatory phase by increasing macrophagic density and Pax7+cells, accelerating the process of myofiber restoration and muscle function.75
Gene therapyAs mentioned above, the anti-ammonia measures utilized in the treatment of HE have limited efficacy. In their trial published in 2015, Torres-Vega et al. employed the Bac-GS baculovirus as a vector for releasing the GS gene. Transduction of the MA 104 or L6 myoblast/myotube cells with Bac-GS produced high expression of the GS gene, with an increase in GS concentration. It was intramuscularly injected into an acute hyperammonemia rat model and the concentration of ammonia decreased 351μM, compared with the controls. A high concentration of GS was detected in the gastrocnemius muscle.76 Those results pave the way for promising options in HE management.
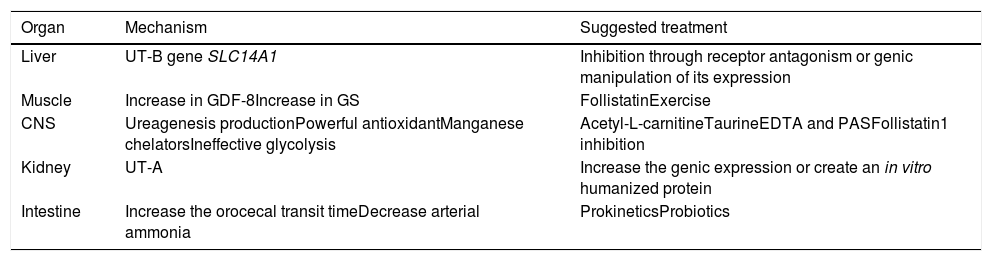
FKS1 inhibitionBecause the FKS1 enzyme regulates glycolysis and impacts lactate production, with the subsequent increased risk for cerebral edema, it could be an interesting therapeutic target to explore. In that respect, low-fructose diets, FKS1 inhibition, and the greater entrance to aerobic metabolic pathways are possible therapies to evaluate in the cirrhotic population (Table 1).
Therapeutic options in HE.
| Organ | Mechanism | Suggested treatment |
|---|---|---|
| Liver | UT-B gene SLC14A1 | Inhibition through receptor antagonism or genic manipulation of its expression |
| Muscle | Increase in GDF-8Increase in GS | FollistatinExercise |
| CNS | Ureagenesis productionPowerful antioxidantManganese chelatorsIneffective glycolysis | Acetyl-L-carnitineTaurineEDTA and PASFollistatin1 inhibition |
| Kidney | UT-A | Increase the genic expression or create an in vitro humanized protein |
| Intestine | Increase the orocecal transit timeDecrease arterial ammonia | ProkineticsProbiotics |
CNS: central nervous system; GDF-8: growth differentiation factor 8; GS: glutamine synthetase; HE: hepatic encephalopathy; UT: urea transporter
The gut microbiota (microbiome) has recently become especially interesting as a therapeutic option in patients with encephalopathy, thanks to the production of inflammatory cytokines generated in the intestine, and the primary production of glutamine and secondary production of ammonia at that site, as well.
It is well known that the human microbiome is a complex of genes that includes more than 1 x 1010 bacteria residing in the gut.75 Said microbiome has widely been characterized as having 5 main phyla: Firmicutes, Bacteroidetes, Actinobacteria, Proteobacteria, and Verrucomicrobia. When those phyla become unbalanced, it is called dysbiosis, which has been extensively described in patients with cirrhosis. In patients with encephalopathy, the most frequent characteristic is the increase in Gram-negative bacteria, especially enterobacteria, which has led to the postulation of fecal transplantation as a treatment option in those patients.76,77
Financial disclosureNo financial support was received in relation to this study/article.
Conflict of interestThe authors declare that there is no conflict of interest.
Please cite this article as: González-Regueiro JA, Higuera-de la Tijera MF, Moreno-Alcántar R, Torre A. Fisiopatología y opciones de tratamiento a futuro en la encefalopatía hepática. Revista de Gastroenterología de México. 2019;84:195–203.

