
The biotechnology-derived medicines known as biosimilars are defined as non-originator treatments that have demonstrated quality, efficacy, and safety comparable to the reference biologic drug. Clinical trials have shown that the infliximab biosimilar, CT-P13, and the candidates for the adalimumab biosimilars, ABP 501 and ZRC 3197, are not significantly different, with respect to efficacy and safety, from the originator drugs in patients with other autoimmune diseases. However, controversy has arisen over the use of biosimilars in inflammatory bowel disease, due to the incipient evidence not only in patients with no previous biotechnology treatment, but also in patients in remission, that could be switched to a biosimilar for non-medical reasons.
The present review is the first critical analysis by different specialists in the area of gastroenterology on the use of biosimilars in inflammatory bowel disease, the evidence on interchangeability, the extrapolation of indications, efficacy, safety, immunogenicity, and the clinical impact of the Mexican health regulations. The aim of our review was to make the positioning and recommendations of these new therapeutic options known, given that they have a potential cost-benefit for both patients and healthcare institutions.
Los medicamentos biotecnológicos biocomparables son definidos como tratamientos no innovadores que han demostrado calidad, eficacia y seguridad comparable al medicamento de referencia. Los estudios clínicos aleatorizados han demostrado que el biocomparable CT-P13 (infliximab) y los candidatos a biocomparables ABP 501 y ZRC 3197 (adalimumab) no difieren significativamente en la eficacia y seguridad respecto al medicamento innovador en pacientes con otras enfermedades autoinmunes. Sin embargo, se ha generado una controversia sobre el uso de los biocomparables en la enfermedad inflamatoria intestinal ante la incipiente evidencia generada no solo en pacientes sin tratamiento biotecnológico previo sino también en remisión y que podrían ser cambiados al biocomparable por razones no médicas.
Esta revisión es el primer análisis crítico de diversos especialistas en el área de la gastroenterología sobre el uso de biocomparables en enfermedad inflamatoria intestinal, de la evidencia de intercambiabilidad, la extrapolación de indicaciones, eficacia, seguridad, inmunogenicidad y del impacto clínico de la regulación sanitaria en México. El objetivo es compartir el posicionamiento y recomendaciones con respecto a estas nuevas opciones terapéuticas que tienen un potencial de costo-beneficio para el paciente y las instituciones de salud.
The rapid development of genetic engineering in the mid-1970s made it possible to produce recombinant proteins in quantities that were sufficient for their therapeutic use. That process incorporated fragments of recombinant DNA, which encoded the production of specific proteins in artificial cell lines that have enabled the creation of biopharmaceuticals used for therapeutic purposes, such as monoclonal antibodies.1
First-generation biotechnology medicines have benefitted millions of patients worldwide in disease treatment and prevention, including inflammatory bowel disease (IBD), certain types of anemia, different cancers, autoimmune diseases, growth deficiencies, reproductive problems, insulin-dependent diabetes, and diverse diseases that have a chronic inflammation component.2 Over the years, biotechnology medicines have shown their efficacy and safety in IBD in numerous clinical studies.
The first biotechnological therapy for Crohn's disease (CD) was approved in 1998 and for ulcerative colitis (UC) in 2006. Infliximab (Remicade®) was the first chimeric monoclonal antibody approved for those indications, followed by the human monoclonal antibody, adalimumab (Humira®), the humanized monoclonal antibody fragment, pegylated certolizumab (Cimzia®), and finally, the human monoclonal antibody, golimumab (Simponi®), for UC. All of them inhibit tumor necrosis factor-α (TNF-α) and have been shown to have an acceptable efficacy and safety profile in clinical studies on IBD.3
After the expiration of the patents of some originator medicines, an intense effort has been made to develop alternative versions known as «biosimilars», defined as non-originator drugs that have demonstrated quality, efficacy, and safety comparable to those of the originators. Biosimilars aim to be structurally identical to the reference medicines, while at the same time offering better cost-benefit to the patient and healthcare systems.4
In Spanish, the terms «biocomparable» and «biosimilar» are synonymous, but the nomenclature had to be changed in the Mexican legislature to avoid confusion for the prescriber and patient with an already existing brand of medicines called biosimilares.
Unlike generic medicines, recombinant proteins, such as monoclonal antibodies, are more structurally complex molecules, which is why there is no «absolute equivalence» between the originator medicine and the biosimilar one: there will always be differences, albeit minimal, that make it necessary for the manufacturer to provide the required studies on biosimilars to demonstrate their quality, efficacy, and safety.
The biopharmaceutical, CT-P13, is the first infliximab biosimilar approved for autoimmune diseases in Europe and the United States, and recently in Mexico.5 Evidence on CT-P13 was provided through drug quality studies and 2 randomized clinical trials, PLANETAS6 (ankylosing spondylitis) and PLANETRA7 (rheumatoid arthritis). The U. S. Food and Drug Administration and the European Medicines Agency also authorized its use in other indications, such as psoriasis, psoriatic arthritis, and IBD, through a regulatory resource called «indication extrapolation», which exempts those diseases from specific clinical studies, taking into account all the evidence demonstrated by the manufacturer in the registration process.8
In Mexico, CT-P13 (infliximab), known as Remsima®, has also recently been approved for all therapeutic indications through indication extrapolation. Thus, Remsima® is authorized to be used in IBD in naïve patients, as well as those in remission. There is growing controversy about extrapolation and interchangeability in relation to biotechnology medicines in Mexico and the rest of the world.
The aim of the present study was to review the available evidence on the efficacy and safety of the interchangeability/substitution of originator biotechnology medicines and biosimilars in IBD. Based on that evidence, we described the position and recommendations regarding their use and discussed the Mexican regulations that apply to those medicines.
Materials and methodsA technical review was carried out on the available evidence with respect to the maintenance of the efficacy and safety of biosimilar medicines in patients with IBD in remission.
Previously, in an academic call for papers for the authors of the present manuscript, several face-to-face sessions were programmed to develop the research. The following keywords, limited to those in English, were defined and searched for in the PubMed® and Ovid®intern international databases: biosimilar ulcerative colitis; biosimilar Crohn's disease; biosimilar inflammatory bowel disease; CT-P13; biosimilar infliximab; biosimilar adalimumab; ABP 501; ZRC 3197; biosimilar interchangeability; biosimilar switch.
Only articles relevant to the aim of the research were selected, based on the following: 1) evidence of a switch from an originator medicine to a biosimilar, specifically in patients with IBD in remission, 2) comprehensive articles with a defined methodology, and 3) a relevant number of patients evaluated (no case series or brief communications). The publications that did not fit those 3 aspects were not considered for the review. Online articles were limited to those published 5 years before and up to August 2017.
The evidence was presented in academic sessions, where it was synthesized, discussed, and agreed upon with 9 of the 10 researchers. No systematic analysis or additional statistics were carried out, nor was the level of quality of the articles methodologically determined.
ResultsA total of 918 articles were found in the databases using the keywords. Only 254 of them were related to biosimilar medicines, with 141 specifically on IBD. A final 57 articles were then under consideration and only 9 met the criteria of the researchers to be reviewed and discussed.
Evidence on the efficacy and safety of ABP 501 (adalimumab) and ZRC 3197 (adalimumab) after the switch was found in relation to autoimmune diseases, but not specifically to IBD.
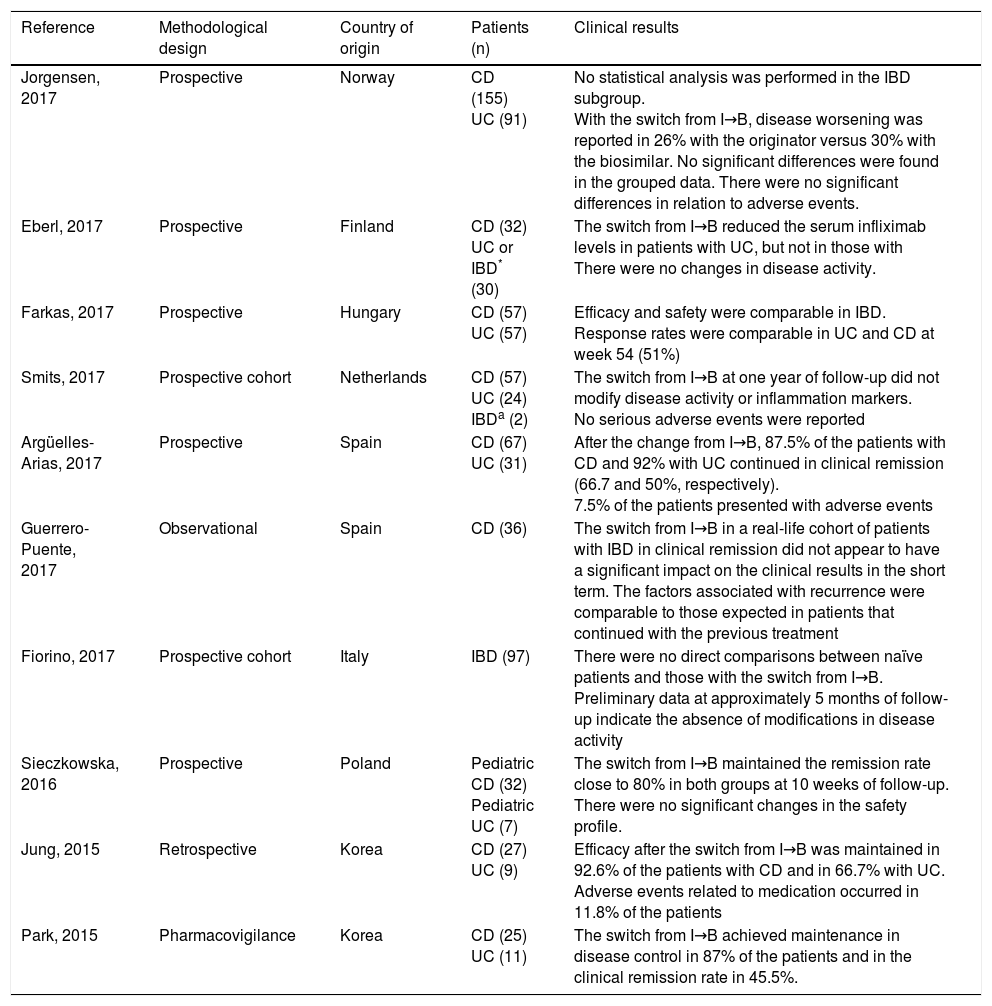
That left a total of 9 phase IV (pharmacovigilance) studies that reported on the maintenance of the efficacy and safety of CT-P13 in patients with IBD in remission. The studies are shown in Table 1 and described below.
CT-P13 interchangeability studies in patients diagnosed with IBD.
| Reference | Methodological design | Country of origin | Patients (n) | Clinical results |
|---|---|---|---|---|
| Jorgensen, 2017 | Prospective | Norway | CD (155) UC (91) | No statistical analysis was performed in the IBD subgroup. With the switch from I→B, disease worsening was reported in 26% with the originator versus 30% with the biosimilar. No significant differences were found in the grouped data. There were no significant differences in relation to adverse events. |
| Eberl, 2017 | Prospective | Finland | CD (32) UC or IBD* (30) | The switch from I→B reduced the serum infliximab levels in patients with UC, but not in those with There were no changes in disease activity. |
| Farkas, 2017 | Prospective | Hungary | CD (57) UC (57) | Efficacy and safety were comparable in IBD. Response rates were comparable in UC and CD at week 54 (51%) |
| Smits, 2017 | Prospective cohort | Netherlands | CD (57) UC (24) IBDa (2) | The switch from I→B at one year of follow-up did not modify disease activity or inflammation markers. No serious adverse events were reported |
| Argüelles-Arias, 2017 | Prospective | Spain | CD (67) UC (31) | After the change from I→B, 87.5% of the patients with CD and 92% with UC continued in clinical remission (66.7 and 50%, respectively). 7.5% of the patients presented with adverse events |
| Guerrero-Puente, 2017 | Observational | Spain | CD (36) | The switch from I→B in a real-life cohort of patients with IBD in clinical remission did not appear to have a significant impact on the clinical results in the short term. The factors associated with recurrence were comparable to those expected in patients that continued with the previous treatment |
| Fiorino, 2017 | Prospective cohort | Italy | IBD (97) | There were no direct comparisons between naïve patients and those with the switch from I→B. Preliminary data at approximately 5 months of follow-up indicate the absence of modifications in disease activity |
| Sieczkowska, 2016 | Prospective | Poland | Pediatric CD (32) Pediatric UC (7) | The switch from I→B maintained the remission rate close to 80% in both groups at 10 weeks of follow-up. There were no significant changes in the safety profile. |
| Jung, 2015 | Retrospective | Korea | CD (27) UC (9) | Efficacy after the switch from I→B was maintained in 92.6% of the patients with CD and in 66.7% with UC. Adverse events related to medication occurred in 11.8% of the patients |
| Park, 2015 | Pharmacovigilance | Korea | CD (25) UC (11) | The switch from I→B achieved maintenance in disease control in 87% of the patients and in the clinical remission rate in 45.5%. |
I→B: the originator drug to the biosimilar drug; n: number of patients
The noninferiority of treatment after the switch from Remicade® infliximab to Remsima® CT-P13, in their different therapeutic indications, was reported by Jorgensen et al. in the NOR-SWITCH study (phase IV): a randomized (ratio 1:1), double-blind trial, with a 52-week follow-up. The primary endpoint was to evaluate disease worsening after switching treatment to the biosimilar, with a noninferiority margin of up to 15%, and the assumption that not more than 30% of the patients would worsen during follow-up. A total of 408 patients with a chronic inflammatory disease (CD, UC, rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, or plaque psoriasis) were included and evaluated for each indication through the respective clinical scales. Statistical analyses in relation to therapeutic indication were not carried out.
The overall results showed that 53 of 202 patients (26%) with IBD worsened with the originator treatment (group not switched to the biosimilar), whereas 61 of 206 patients (30%) worsened with CT-P13 (group switched to the biosimilar). There were no significant differences in the overall statistical analysis or in the frequency of serious adverse events (close to 10%) or immunogenicity.9
In addition, Eberl et al. reported the absence of significant differences in the percentage of anti-infliximab antibodies after the switch in therapy. However, a greater number of patients with UC presented with significantly lower serum levels of infliximab after the switch. That pharmacologic phenomenon was not observed in patients with CD.10
Clinical experience with CT-P13 was also reported by Farkas et al. in a study with a 54-week follow-up. Patients with CD and UC were monitored to evaluate the maintenance of remission after the switch in therapy. The clinical response rate in the two groups (51%) was comparable to the rates reached at week 14 (65.5 and 75.5%, respectively).11
Smits et al. reported the absence of significant changes in the disease activity scale and inflammatory markers in IBD during one year of follow-up in 83 patients after the switch. Only 7% of the patients interrupted therapy with CT-P13 due to adverse events. Moreover, 8% of the patients were positive for anti-infliximab antibodies.12
Similar results were found by Jung et al. in a retrospective multicenter study with 54 weeks of follow-up on patients with CD (n = 27) and UC (n = 9). A total of 92.5% of those patients maintained the clinical response rate achieved with the previous treatment.13
Argüelles-Arias et al. recently demonstrated the effectiveness and safety of the switch to CT-P13 in 98 patients with IBD at 6 months of follow-up. The remission achieved with the previous treatment was maintained in both CD and UC (66.7 and 50%, respectively). There were no significant differences in the safety profile after the switch.14
In the post-marketing study by Park et al., those authors reported a total of 87% of patients with disease control, 45.5% of whom reached or maintained clinical remission.15
Thirty-one Italian hospital centers participated in the pharmacovigilance PROSIT-BIO study by Fiorino et al. The researchers underlined relevant data with respect to the maintenance of efficacy and safety in 313 patients with CD and in 234 with UC, 97 of whom had been switched to CT-P13. No disease activity modifications were found at 5 months of follow-up.16
In another observational study, Guerrero-Puente et al. evaluated the efficacy, safety, bioavailability, and relapse-associated factors after the switch to the biosimilar. Thirty-six patients were selected and no significant differences in the short-term clinical response or modifications in the serum concentration of the biopharmaceutical were found.17
Finally, not much information on the pediatric population was found: only the study by Sieczkowska et al. stood out. They showed results with respect to switching to the biosimilar in pediatric patients with IBD. Three hospitals took part in that prospective study: a total of 32 patients with CD and 7 with UC were selected. Age ranged from 3 to 15 years, with a mean of 12 years. At 10 weeks of monitoring, maintenance of remission was conserved in 80% of the patients, with no significant modifications after the treatment change. The CT-P13 safety profile was consistent with that reported for the reference medicine.18
DiscussionThe aim of the present study was to review the available evidence on the maintenance of the efficacy and safety of biosimilar medicines in patients with IBD in remission. Previous studies on the efficacy and safety in naïve patients have been described.19 However, there is much debate about the evidence on maintenance after the switch from the biologic originator medicine to the biosimilar, as well as the lack of health regulations with interchangeability among biotechnology medicines. To the best of our knowledge, ours is the first article on that theme in Mexico.
Because the candidates for adalimumab biosimilars, ABP 501 and ZRC 3197, are not yet sold in Mexico, and no studies on them were found, we could not discuss their use in the present study.
In general, clinical experience with the infliximab biosimilar, CT-P13 (Remsima®), tends to show positive results that support its use in IBD. According to results, the change of therapy does not appear to modify the inflammatory markers of the disease, nor does it negatively impact the maintenance of remission achieved with previous treatment. Nevertheless, it must be kept in mind that in Mexico there is still not enough clinical experience, partially due to the only recent approval of Remsima®, making it necessary to gain experience and determine the economic and social burden of those diseases on both the patient and the healthcare systems.
We found that there is little available information and the methodological criteria of the studies that do exist are heterogeneous, which is understandable in part, given that the analyses are phase IV (pharmacovigilance) studies and there are no initial trials conducted on CT-P13 in IBD. Therefore, we were unable to determine the level of evidence through a systematic analysis or additional statistical analysis (i.e., a meta-analysis), because our study is a literature review to familiarize the specialist with the subject of biosimilars. Further analyses are needed to broaden the evidence.
Notwithstanding, the medical implications of introducing biosimilars to IBD are described below.
Legal framework in MexicoIn article 222-Bis of the Regulations on Health Inputs of the General Health Law of Mexico, a biosimilar medicine is defined as a non-originator medicine that has been shown to have comparable quality, efficacy, and safety to those of the reference medicine.20 The registration of a biosimilar medicine by the Health Department is also based in part on the Mexican Official Norms 257, 177, and 220, which describe the technical requirements for demonstrating biosimilarity.21–23
We did not find a document or official guide that defined the terms and specific criteria for the interchangeability and extrapolation between biotechnology medicines, which potentially adds the element of uncertainty to their performance.
InterchangeabilityInterchangeability is the substitution of one medicine for another of the same pharmaceutical composition, that is expected to produce the same therapeutic effect and maintain the efficacy and safety in stabilized patients that was achieved with the previous treatment.24
Interchangeability between biotechnology medicines is a controversial subject. In the United States, the Food and Drug Administration has officially stated that interchangeability is the responsibility of each of the States and remains neutral on the matter. Furthermore, the European Medicines Agency makes no recommendations, nor does it require interchangeability studies for the registration of biosimilars with the health authority, leaving the decision up to each member state of the European Union, physician, or pharmacist.25
We found no clear definition in the Mexican Norm of the process for demonstrating interchangeability between the originator drug and the biosimilar. In contrast, in relation to generic medicines, the General Healthcare Council, through a commission of experts on interchangeability testing, determined a series of methodological requirements to demonstrate said quality between medicines with the same qualitative-quantitative composition.22 However, the health regulation for generic medicines is not applicable to biosimilars, given the clear pharmacologic differences between them and their different production processes.1,2
Furthermore, in the majority of cases, a biotechnology medicine is acquired through a public agency, given its high cost and pharmaceutical control. In that setting, both the originator medicine and the biosimilar legally share the same health sector code.20 No distinction is made between them, which is why a healthcare institution can promote the change of therapy without the consent of the patient or the physician or evidence of its efficacy. That leaves an important void in the actual traceability of treatment and efficiency in pharmacovigilance. We found no valid motive for changing the biotechnological therapy of a patient that is in remission, thanks to a previous therapy, other than the cost of the treatment. In our opinion, that practice is a breach of bioethical principles in medicine and of the human right to health protection.
Nevertheless, we recognize that the advantages of sharing the health sector code lie in the greater price competitiveness of this type of product that potentially will make it more accessible to the patient and minimize the shortage of medications. In that sense, the change from originator treatment to a biosimilar is debatable.
The Food and Drug Administration recently published a general guideline for evaluating the interchangeability of biotechnology products. It recommends carrying out prospective studies that assess the noninferiority of treatment in at least 2 formulation changes.26
The phase IV NOR-SWITCH study was specifically designed to demonstrate the interchangeability between the originator drug, infliximab, and the biosimilar, CT-P13 in all therapeutic indications. It is an independent trial that is not funded by the drug manufacturer. Even though the authors did not carry out a subgroup comparison, we carefully analyzed the data and found that 14 of 78 patients (17.9%) with CD worsened with the originator treatment (group not switched to the biosimilar) and 23 of 77 patients (29.8%) worsened with CT-P13 (group switched to the biosimilar) within a 54-week follow-up period. Likewise, 3 out of 47 patients (6.3%) with UC worsened with the originator treatment (group not switched) and 5 out of 46 patients (10.8%) worsened with CT-P13 (group switched to the biosimilar). More statistical sub-analyses will provide evidence on the consistency of that study's conclusions.
We found the results of the NOR-SWITCH9 study to be controversial. For example, 1) in the randomization of the patients, neither the disease phenotype nor the definition of «stable» was considered, which was reflected in a greater bias in the result conclusions; 2) the patients in endoscopic remission were not defined; 3) the statistical results of noninferiority showed a high degree of variability in the clinical response in each population subgroup, but not in the total patient group; 4) it should be pointed out that the response in IBD was less variable, compared with those of other populations, but making disease worsening the primary endpoint was a debatable decision, given that noninferiority refers to a statistical term and worsening to a clinical term; 5) clinical scales were used to evaluate disease activity and endoscopic scales were not considered; and 6) no subgroup stratification of the safety data was presented after the switch.
A study considering those variables would contribute a greater level of evidence and solidity for demonstrating interchangeability between originator infliximab and biosimilar infliximab. The results of the present NOR-SWITCH study are still inconclusive for demonstrating interchangeability and we must wait for the results of its extension phase.
ImmunogenicityThe process of immunogenicity, or the production of anti-drug antibodies, is clinically relevant, because those antibodies have the potential to modify the therapeutic response or certain pharmacokinetic-pharmacodynamic aspects. At present, we have special laboratory tests for measuring the level of anti-drug antibodies (i.e., the ELISA test). Unfortunately, their routine measurement in clinical practice is not often performed, due to the lack of that technology in our hospitals. The heterogeneity in the standardization of laboratory test methodology, low sensitivity for the biopharmaceutical, and the great variability of results reported in the literature are other factors limiting its frequent use.
Immunogenicity has been shown to largely depend on the degree of genetic «humanization» of the recombinant protein,27 regardless of whether it is an originator or a biosimilar, while at the same time biopharmaceuticals are similar in their structure and consistently conserve the quality of the medicine from batch to batch.28
In theory, biosimilars should not produce greater immunogenicity compared with the originator, because they conserve the same immunodominant epitopes, which are regions of amino acids that act as antigenic determinants with the capacity to produce antibodies. Ben-Horin et al. demonstrated that the systemic anti-infliximab antibodies (polyclonal) that circulate in the patient are not different and they recognize the same molecule, infliximab.29
Baert et al. also reported that the systemic presence of anti-infliximab antibodies increased the risk for adverse reactions during infusion and was related to the loss of response in patients with IBD.30 Likewise, Farrel et al. observed that patients in remission had a lower serum concentration of anti-infliximab antibodies, similar to immunosuppressed patients with premedication, in contrast to those that lost clinical response.31
There is little information on patients previously sensitized to some type of biotechnology therapy. Bálint et al. found that those patients developed peri-infusional adverse reactions and presented with a significant increase in the serum concentration of anti-infliximab antibodies, compared with naïve patients.32 Recent studies have confirmed the same findings and have been thoroughly described by Hindryckx et al.33
Based on the above, in the exceptional case of an increase in immunogenicity from the change in therapy, we propose that there is a pathophysiologic mechanism not yet completely understood, through which the immune system «loses its tolerance» in relation to the biosimilar drug, and thus the change to a biosimilar could be a risk factor. We recommend, when possible, the measuring of anti-drug antibodies before and after the switch, within a reasonable time frame, to rule out variables that would affect clinical remission resulting from the treatment in previously sensitized patients. Other factors that should be considered are premedication/comedication with immunosuppressants, non-biotechnology medicine concomitance, the treatment optimization strategy, the heterogeneous glycosylation profile of the biosimilar in relation to the originator, and possible inconsistencies in the drug manufacturing quality.
In addition, the subject of opportunistic infections that appear due to TNF-α inhibition in long-term treatment should be comprehensively studied in clinical trials, given that problems have been reported during intravenous infusion or derived from the use of a medical device for applying subcutaneous doses.34 There are few studies on this topic conducted on biosimilars.
Finally, the long-term safety profile can be a key differentiator at the time of interchangeability between biotechnology medicines. For example, in the extension phase of the PLANETAS study (ankylosing spondylitis) at 102 weeks, treatment was reported as noninferior after the switch to CT-P13, the same as occurred in the extension phase of the PLANETRA study (rheumatoid arthritis).35,36 It should be pointed out that the frequency of adverse events in patients with ankylosing spondylitis that were switched to the biosimilar was 71.4%, whereas it was 48.9% in the naïve patients that began treatment with CT-P13. That was a difference of 22.5% between groups treated with the same CT-P13, which could support our recommendation of producing specific evidence for IBD, given that the clinical variables in patients in remission and naïve patients are different, and cannot necessarily be extrapolated.
Indication extrapolation in inflammatory bowel diseaseIndication extrapolation has been defined as the regulatory resource that exempts the biotechnology medicine from clinical studies in determined therapeutic indications to obtain permission for commercialization, taking into account all the evidence demonstrated by the manufacturer during the registration process, in terms of quality, efficacy, and safety in the main indications.8
In Mexico, extrapolation is decided upon in the regulatory setting after a «case-by-case» evaluation of the factors related to the biochemical structure and function of a biosimilar, based on the opinion of expert committees of the Federal Commission for the Protection against Health Risks (COFEPRIS, the Spanish acronym).37
The technical and scientific criteria by which the health agencies grant extrapolation are: 1) that both biopharmaceuticals have the same pharmacologic mechanism; 2) that they have the same administration route and pharmaceutical composition; 3) that there is linear elimination of the biopharmaceutical (dose-dependent); 4) that pharmacokinetic bioequivalence studies are presented; 5) that clinical studies in sufficiently sensitive models are presented; and 6) that not only immunogenicity, but also the safety profile, is solidly characterized, among other aspects.38–41
However, there is evidence that the pharmacologic mechanism of action and the pharmacokinetic behavior is not the same in all anti-TNF-α agents,42 which could partially explain the clinical differences found in clinical practice. This gives pause for serious reflection on the clinical impact of indication extrapolation in IBD, given that the mechanism is not strictly the same, and at the time, was not broadly characterized by the originator drug manufacturer.
Nevertheless, we discussed the potential benefits of extrapolation and concluded that they include several aspects: 1) not exposing the patient to unnecessary risks that could come out of a redundant study, implying a bioethical debate on the benefit of clinical research; 2) the difficulty of conducting a solid study, given the limited sample size in IBD; 3) study financing, which would raise the cost of treatment; and 4) the stimulation of economic competition, favoring treatment access for a larger number of patients through the expedited approval of biosimilars.
We concluded that extrapolation brings about uncertainty, because the regulatory resource oversees the evaluation of the medicine and not the clinical variables associated with the disease. Those variables importantly modify the response of the drug and are not considered in the clinical studies that are needed to support the prescription of the medicine. Thus, the physician is at a disadvantage in his or her medical practice because decisions made by the specialist are always based on clinical evidence and on the experience gained from the use of the previous medicine.
Obtaining the generalized medical acceptance of indication extrapolation as the regulatory resource will not be an easy task for the manufacturers of biosimilars, in relation to IBD, as long as adequate channels of communication are not pursued that go beyond the normative aspects of medication authorization that do not always relate to the prescriber or the patient.
Position and recommendations- 1.
We support the introduction of biosimilar medicine into inflammatory bowel disease in Mexico.
- 2.
However, biosimilarity continues to be a fairly uncommunicated theme in the medical community of IBD specialists.
- 3.
Even though there is no epidemiologic IBD register in Mexico, national reference centers have observed a trend toward an increasing number of cases, which implies a greater need for biotechnology therapies or new treatment options. Biosimilar medicines are a therapeutic alternative that can provide greater access to patients with IBD, along with its consequent benefits.
- 4.
Because IBD has variables that are clinically different from other autoimmune inflammatory diseases, we recommend conducting specific clinical studies on the indication so that the use of biosimilars can be recommended with greater accuracy and that patients seeking medical consultation can be given maximum information.
- 5.
Currently, the specific evidence for extrapolating a biosimilar drug from rheumatic disease to IBD is not clearly defined, thus its recommendation should not be generalized. We would agree upon indication extrapolation, only if the evidence has been previously defined through an official consensus, or the health agencies or the biosimilar manufacturer communicate the criteria through which commercialization was authorized.
- 6.
Mexico is a regulatory leader in biosimilar drug evaluation, but the criteria for the extrapolation of rheumatic diseases to IBD are not clear, and therefore more evidence is required for its recommendation.
- 7.
The lack of a clear definition of interchangeability between biotechnology drugs in Mexico is a legal void that makes the clinical concept appear to be an administrative subject and does not take into account the medical and scientific evidence necessary for making accurate patient prognosis.
- 8.
We are in favor of the interchangeability between biotechnology products, as long as the decision is agreed upon between the physician and the patient and it is supported by the maximum level of scientific evidence.
- 9.
We do not recommend interchangeability or automatic substitution for nonmedical indications, due to the potential risk for
- 10.
We recommend that the manufacturers of biosimilars conduct interchangeability studies specifically on IBD starting from the initial clinical development.
- 11.
We encourage the community of gastroenterologists and specialists in IBD, as well as other healthcare professionals, to report all the adverse events and lack of efficacy of all medicines, for the promotion of a culture of pharmacovigilance in clinical practice.
The authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Financial disclosureNo financial support was received in relation to this study/article.
Conflict of interestThe position expressed by the experts is unanimous and does not necessarily reflect the view of the institutions where they work.
C.M. del Real, B. Rubio, and M. Rojas declare that they received an academic grant from Abbvie, Pfizer, Janssen, and UCB to attend at least one medical congress. Y. Gutiérrez declares she received fees from Takeda for giving conferences and an academic grant from Abbvie to attend at least one medical congress. X. Sánchez declares that she received an academic grant from Janssen to attend at least one medical congress.
A. Mayoral declares having received fees from Abbvie, Pfizer, and Janssen for giving conferences and an academic grant from Abbvie, Pfizer, and UCB to attend medical congresses, as well as from Janssen for brand consulting services. A. Esquivel declares having received professional fees from Abbvie and Pfizer for giving conferences and for brand consulting services. J.L. Rocha declares having received grants from Abbvie, Pfizer, Janssen, and UCB to attend at least one medical congress and professional fees from Abbvie, Pfizer, and Janssen for giving conferences. Finally, J. Yamamoto declares having received fees from Abbvie, Pfizer, Janssen, and UCB for giving conferences and an academic grant from Abbvie to attend at least one medical congress, as well as professional fees from Bristol for having participated in a clinical study. J. Ramos declares that he has no conflict of interest.
The authors wish to thank the physicians, Azucena Casanova and Alejandro García Martínez for their valuable help in the preparation of the manuscript.
Please cite this article as: Mayoral-Zavala A, Esquivel-Aguilar A, del Real-Calzada CM, Gutiérrez-Grobe Y, Ramos-García J, Rocha-Ramírez JL, et al. Actualización sobre los medicamentos biocomparables en la enfermedad inflamatoria intestinal: posición y recomendación en México. Revista de Gastroenterología de México. 2018;83:414–423.

