


La granulomatosis con poliangitis (GPA), anteriormente granulomatosis de Wegener1,2, es una vasculitis necrotizante de pequeños vasos, con inflamación granulomatosa sistémica predominantemente en el tracto respiratorio alto, pulmones y riñones; asociada a la presencia de anticuerpos contra citoplasma de neutrófilos (cANCA) y anticuerpos anti-proteinasa 3 (anti-PR3)1–3. Aunque infrecuente, se han reportado casos de GPA con afectación gastrointestinal (GI) en un 5-11%1,2. Las manifestaciones clínicas varían desde dolor abdominal hasta sangrado masivo y perforación3,4.
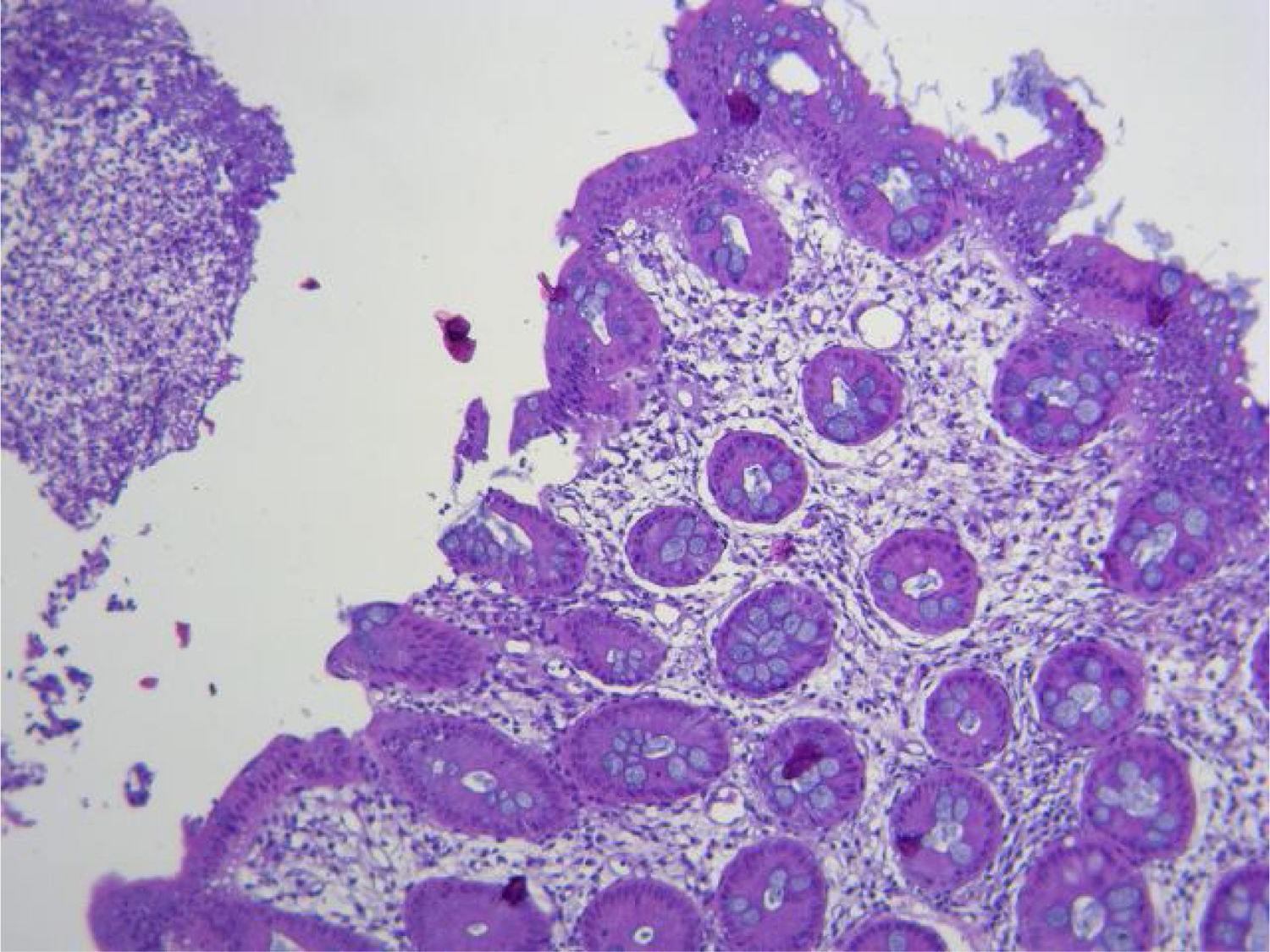
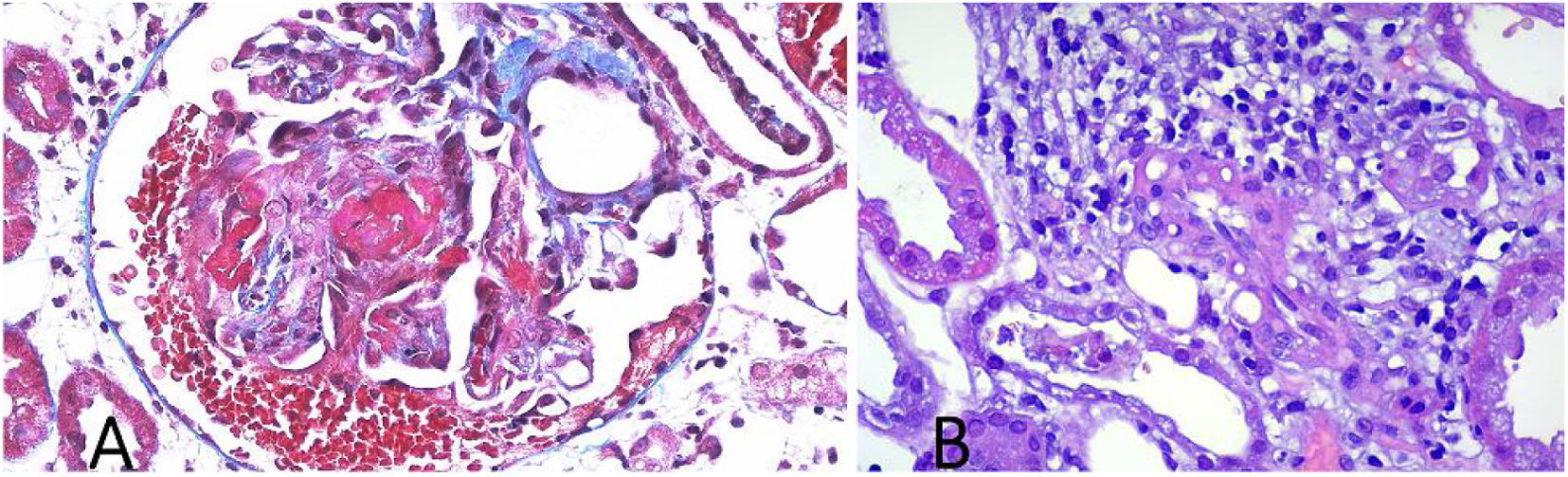
Presentamos el caso de un femenino de 57 años, sin antecedentes relevantes, con un cuadro de dos meses con síntomas constitutivos y poliartralgias simétricas, agregándose evacuaciones melénicas en tres ocasiones, por lo que acude a Urgencias. A su ingreso, con signos vitales dentro de parámetros normales, exploración física con dolor abdominal en hipogastrio, peristalsis aumentada, sin datos de irritación peritoneal, flogosis articular en muñecas y tobillos, llenado capilar aumentado, resto sin alteraciones. Se solicitan exámenes de laboratorio reportaron hemoglobina 9 g/dL, leucocitos 13,220 cel/mm3, PMN 81.3%, creatinina 3.40 mg/dL, BUN 39.5 mg/dL, urea 84.6 mg/dL, depuración creatinina 14.6 mL/min, sodio 135 mmol/L, potasio 4.99 mmol/L, proteína C reactiva 210 mg/dL, EGO: proteinuria 1.51 g/L, hemoglobinuria +++, > 100 eritrocitos, 8-10 leucocitos por campo y urocultivo negativo. Se realiza tomografía computarizada (TAC) simple de abdomen reportando engrosamiento mural y edema de mucosa gástrica y en íleon terminal, con escaso líquido libre. Se inicia manejo con omeprazol y antibioterapia con linezolid y metronidazol. Se realizan panendoscopia y colonoscopia observando úlcera de 5 mm en antro, Forrest III, íleon terminal y ciego con múltiples ulceraciones pequeñas con fibrina y patrón empedrado, se toman biopsias reportando infiltrado inflamatorio perivascular, exudado fibrinoso, edema de lámina propia, sin evidencia de malignidad. (fig. 1). Se inicia metilprednisolona y mesalazina. Se interconsulta con reumatología, se evidencia C-ANCA positivo 1:1,280 y anti-PR3 positivo con 427 IAs. Se toma biopsia percutánea renal reportando granulomatosis con poliangitis (fig. 2). Se agrega ciclofosfamida y profilaxis contra Pneumocystis jirovecii con trimetoprima/sulfametoxazol. La paciente presenta mejoría y se egresa al séptimo día de hospitalización. El seguimiento a seis meses sin eventualidades.
Actualmente la GPA tiene una incidencia mundial de 20 casos por millón, afecta edades entre los 50-70 años, con predominio en población caucásica y asiática2,4. Dentro de las manifestaciones clínicas se encuentran síntomas constitucionales, glomerulonefritis necrotizante rápidamente progresiva, afectación de tracto respiratorio, nervios periféricos, ojos, articulaciones y piel1,2,4. La afección GI es rara, suele presentarse después del diagnóstico de la GPA, cuando ocurre simultáneamente, se asocia a mayor afectación renal y mortalidad1,2,5. Es causada por una vasculitis mesentérica o un daño directo a los vasos de la submucosa2–6. Suele presentarse como dolor abdominal, náusea, vómito, sangrado tubo digestivo, isquemia intestinal y perforación3,5–8. Estos síntomas suelen remitir con el manejo de la GPA1,2. Sin embargo, es necesario excluir otras causas de afección gastrointestinal antes de iniciar tratamiento1,6,9.
El diagnóstico de la afección GI en la GPA se basa en la presencia de síntomas GI asociados con afección a otros órganos, y se complementa con la presencia de C-ANCA en 90%, anti-PR3 en 75% o mieloperoxidasa en 20%2,4,6–8. En los estudios de imagen puede observarse neumoperitoneo, engrosamiento mural, abscesos, líquido libre y microaneurismas en la arteria mesentérica1,9. La endoscopia y colonoscopia pueden ser normales, inclusive en casos severos, sin embargo, comúnmente se observan múltiples úlceras pequeñas, redondeadas, de fondo claro asociado a coágulos o sangrado reciente, con eritema y edema difusos de mucosa1,5,9. Las biopsias confirman el involucro GI, reportando infiltrados de neutrófilos en los pequeños vasos (arteriolas, vénulas y capilares) submucosos causando necrosis, ulceración de la mucosa, inflamación granulomatosa rodeada de necrosis fibrinoide irregular, trombosis y fibrosis1,4,5,9.
El manejo conservador con ayuno, fluidoterapia, nutrición, antibioterapia sistémica y glucocorticoides sistémicos, en combinación con inmunosupresores como ciclofosfamida o rituximab, permite la resolución en semanas de la mayoría de los pacientes1,2,4. En los casos de perforación intestinal o sangrado severo, se requiere manejo quirúrgico1,3,6,8.
El pronóstico a seis meses sin tratamiento es fatal4. Con tratamiento, el pronóstico es reservado, por la alta tasa de relapso (30-60%)2,4,7. Las principales causas de muerte son las complicaciones quirúrgicas en un 44%7 y la falla renal crónica (50%)2.
En nuestro conocimiento este es el primer caso reportado en América Latina de una paciente con GPA con afectación GI, es importante hacer hincapié en la relación entre la GPA y el sangrado de tubo digestivo para el diagnóstico y tratamiento oportuno.
Consideraciones ÉticasEste estudio se realizó cumpliendo la normativa actual en investigación bioética conformado por los siguientes criterios como la protección de personas y animales, confidencialidad de los datos siguiendo los protocolos del centro de trabajo sobre su publicación y preservación de su anonimato, derecho a la privacidad y consentimiento informado. Los autores declaran que no existe información personal que permita identificar a la paciente. Este estudio no recibió ningún apoyo específico por parte de agencias de fondos, en el sector público, comercial o con fines de lucro. Los autores declaran no tener conflicto de intereses en la redacción de este manuscrito.
FinanciaciónEste trabajo no ha recibido ningún tipo de financiación.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

