



El síndrome de Allgrove (SA) es un trastorno neuroendocrino progresivo de etiología desconocida, herencia autosómica recesiva, caracterizado por acalasia, alacrimia, insuficiencia adrenal, incluso alteraciones autonómicas y neurológicas; con mutaciones homocigóticas en el gen AAAS localizado en el cromosoma 12q13 que codifica la proteína ALADIN (la mutación más reportada a nivel mundial es la c.1331+1G>A), y prevalencia estimada en 1/1,000,000 personas1,2.
Paciente femenina, de 1 año 9 meses de edad, padres y hermano mayor sanos, no consanguinidad; segunda gesta a término, peso 3.075kg, longitud 48cm, no hipoxia perinatal. Desde su nacimiento, ausencia de lágrimas; desnutrición (ganancia de 900g en 18 meses); vómito desde los 9 meses de edad (diario, abundante, posprandial inmediato de alimentos no digeridos) y disfagia a sólidos (ingesta de 65kcal/kg/día). Recibió sin mejoría procinético, inhibidor de bomba de protones (IBP) y fórmula extensamente hidrolizada.
El examen físico mostró: peso 6.2kg, longitud 74cm, perímetro cefálico 43cm, sin fascies características, alopecia parcial, sin lágrimas, atrofia y debilidad muscular generalizada, retraso global del desarrollo psicomotor (RGDP) y desnutrición crónica grave. No se observó hiperpigmentación cutánea.
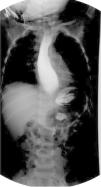
Se admitió para recuperación nutricional con alimentación enteral de acuerdo con las directrices de la Organización Mundial de la Salud para el tratamiento hospitalario de niños con desnutrición grave. La serie esofagogastroduodenal (SEGD) mostró imagen en «pico de ave» (fig. 1). La endoscopia evidenció dilatación esofágica distal con residuos alimentarios. La manometría esofágica de alta resolución concluyó acalasia esofágica (AE) tipo i (fig. 2). La prueba de Schirmer fue positiva y los niveles de hormona adenocortipotropa (ACTH) y cortisol, normales.
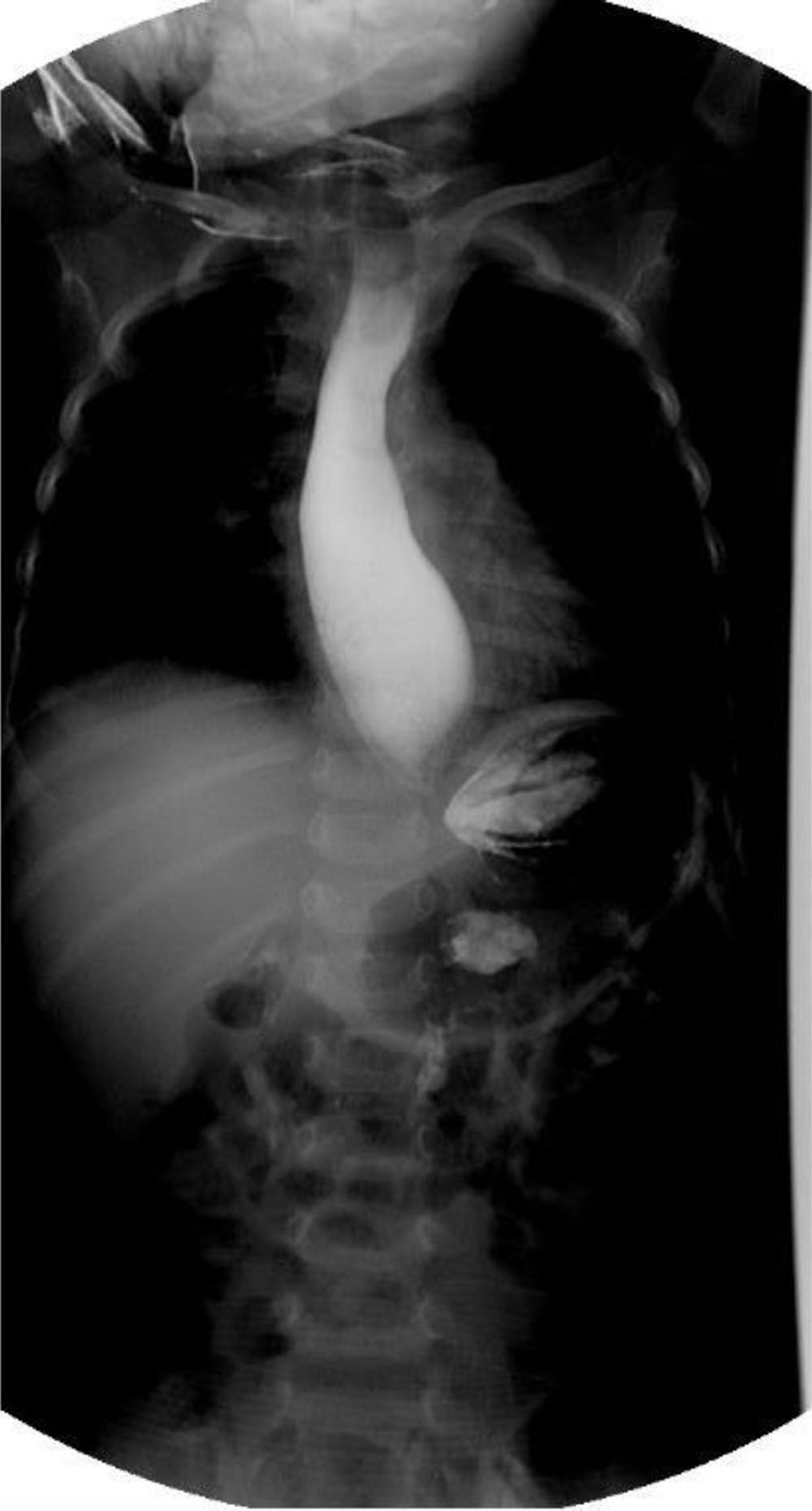
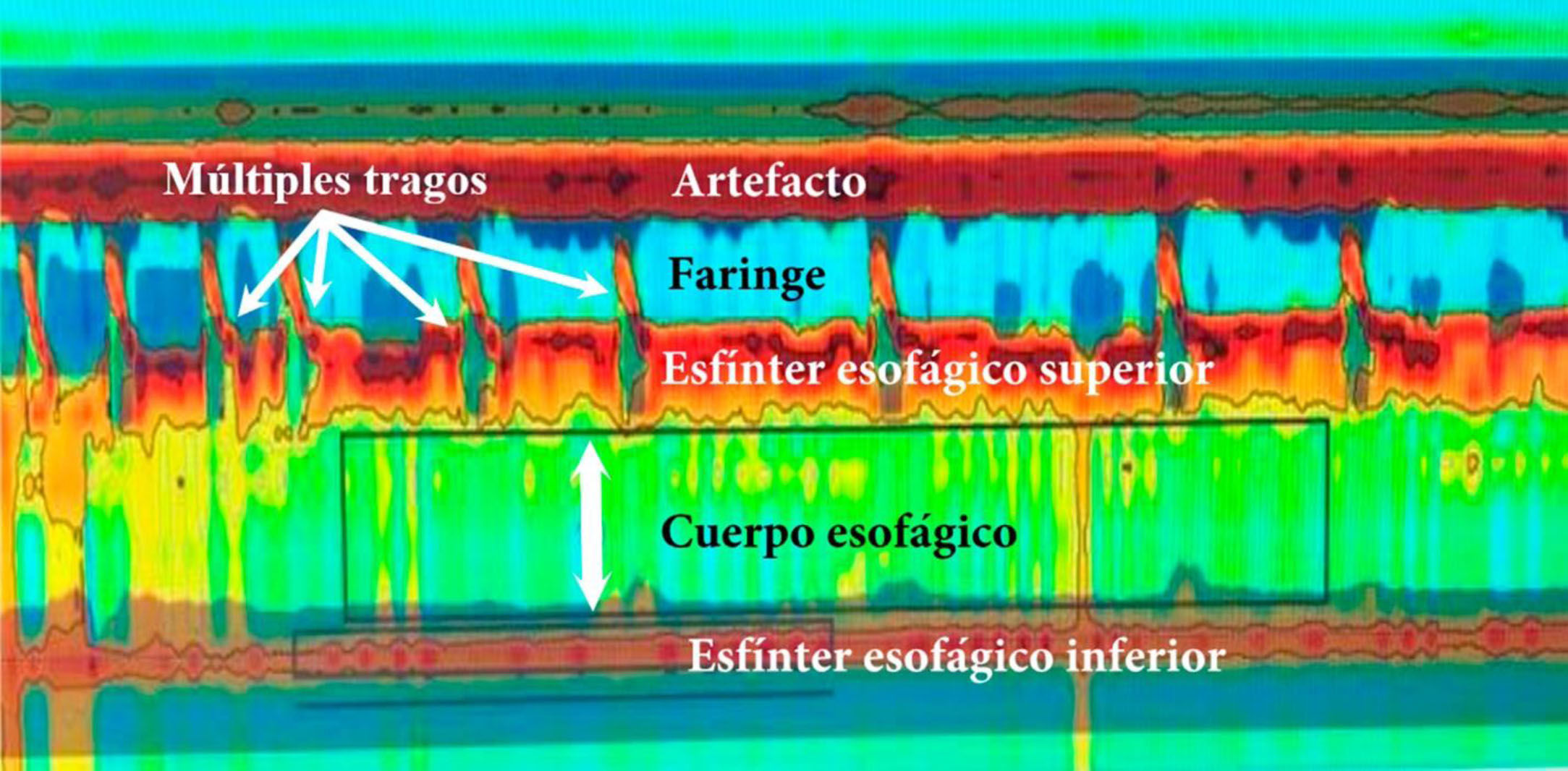
Manometría esofágica de alta resolución. Acalasia tipo i. Nótese la coordinación entre la contracción faríngea y la relajación del esfínter esofágico superior con cada trago y la ausencia tanto de peristalsis del cuerpo esofágico como la relajación del EEI. PIR media 22mmHg (normal <20), ausencia de peristalsis de cuerpo esofágico en 100% de las degluciones, ausencia de relajación del EEI, presión media del EEI 48mmHg (normal 10-45); ICD medio 160mmHg/cm/s (normal 450-8,000).
EEI: esfínter esofágico inferior; ICD: integral de contractilidad distal; PIR: presión integrada de relajación.
Fue sometida, sin complicaciones, a una cardiomiotomía de Heller con funduplicatura parcial anterior tipo Dor por vía laparoscópica; 14 meses después presentó disfagia a sólidos; se observó ligera dilatación esofágica distal y se dilató con balón hidroneumático a 10mm a nivel del esfínter esofágico inferior (EEI).
Actualmente, con 4 años 2 meses de edad, presenta adecuada distensibilidad esofágica, sin estenosis y pliegue quirúrgico funcional; mejoría nutricional (desnutrición crónica leve) con velocidad de crecimiento 6.5cm/año; sin defecto epitelial corneal y sin insuficiencia adrenal. Recibe terapia física y ocupacional por RGDP, bajo tratamiento procinético y dieta licuada artesanal por regurgitaciones.
Existen pocos reportes pediátricos en Latinoamérica. Este se trataría del primer reporte de SA en lactante latinoamericana. De lo publicado en México, la alacrimia se presentó como primer síntoma, seguido de acalasia; la insuficiencia adrenal se documentó a los 5 años de edad en uno de 3pacientes; otro cursó con disfunción neurológica periférica y autonómica3-5.
Los recién nacidos a término presentan lágrimas desde el primer día de vida, producción desarrollada completamente entre la 1-7 semanas de vida. La alacrimia se considera un síntoma temprano del SA, lo que debe conducir a considerarlo como diagnóstico diferencial6.
La AE es un trastorno de motilidad con prevalencia anual estimada en 0.18/100,000 niños, cursa con vómito, disfagia y pérdida ponderal, síntomas documentados en nuestra paciente secundarios a AE y derivando en desnutrición grave. El 46.1% de los casos presenta tos crónica, que suele confundirse con enfermedad por reflujo gastroesofágico, y hasta la mitad recibe procinéticos e IBP antes del diagnóstico, como ocurrió en este escenario7. El Colegio Americano de Gastroenterología establece que debe sospecharse en pacientes con disfagia a líquidos y sólidos, y en aquellos con regurgitaciones que no respondan a prueba terapéutica con IBP a dosis adecuada8. La SEGD como imagen de primera línea apoya el diagnóstico de AE, pero puede resultar negativa hasta en un tercio de los casos. Es útil para evaluar cambios tardíos que tienen implicaciones en el tratamiento. Se recomienda a todos los pacientes evaluación endoscópica de la unión gastroesofágica para descartar obstrucción mecánica o seudoacalasia y aquellos sin evidencia de obstrucción deben someterse a manometría convencional o de alta resolución, esta última estándar de oro para diagnóstico, la cual no solo ha mejorado la capacidad diagnóstica, sino también nuestra capacidad para guiar el tratamiento en el tipo de AE8. Existen abordajes terapéuticos para AE enfocados en reducir la presión del EEI y facilitar el vaciamiento gástrico. Hay medidas con efecto temporal como aplicación de toxina botulínica y, para pacientes con alto riesgo quirúrgico (que no son candidatos a anestesia), administración por vía oral de bloqueadores de canales de calcio, y tratamientos de efecto prolongado como dilatación esofágica y esofagomiotomía (con o sin funduplicatura)9. El tratamiento de elección para la AE es todavía controversial, heterogéneo e involucra a un equipo multidisciplinario; muchos recomiendan la miotomía tipo Heller vía laparoscópica y otros, la dilatación esofágica, en función del subtipo de AE10. En general, se recomienda discutir las opciones con la familia, tomando en consideración la experiencia del centro donde se trata al paciente. No existen muchos estudios pediátricos que reporten los resultados de intervenciones de efecto duradero.
La crisis adrenal es la primera causa de mortalidad en el SA, ocurre como episodios de hipoglucemia e hiperpigmentación progresiva, y se limita mayormente a deficiencia de glucocorticoides11. Es importante destacar que la insuficiencia suprarrenal resistente a ACTH como parte de la tríada clásica es evidente a fines de la primera década de vida, pudiendo presentarse 5-10 años posteriores al inicio de síntomas de AE, desarrollarse durante la infancia tardía, adolescencia e incluso edad adulta12,13. Por lo anterior, se sugiere monitorización continua de electrolitos séricos, especialmente durante eventos de estrés11.
Desafortunadamente, nuestra limitante es que no fue posible obtener el estudio genético por razones económicas, que refleja la práctica clínica real. Esta enfermedad es genéticamente muy heterogénea y existen casos descritos de pacientes diagnosticados sin mutaciones en el gen AAAS, por lo que pudiera haber otros genes implicados2. Sin embargo, el diagnóstico de acalasia está confirmado, la paciente está recibiendo el tratamiento adecuado a pesar de su temprana edad y consideramos que nuestro reporte ayudará a otros a identificar estos casos para evitar retrasos en el diagnóstico, sobre todo en niños menores de 2 años de edad que inician con síntomas inespecíficos.
Por lo raro de esta condición, debemos tener alto índice de sospecha, enfatizar en una historia clínica adecuada y conocer el abordaje de uno de los síntomas más frecuentes en población pediátrica.
Responsabilidades éticasLos autores declaran que este artículo no contiene información personal que permita identificar al paciente. Debido a que se realiza revisión de expediente clínico no amerita autorización del Comité de Ética.
FinanciaciónEl presente trabajo no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.
Conflicto de interesesLos autores declaran no haber tenido ningún tipo de conflicto de intereses.
Dra. Alejandra Consuelo Sánchez, Departamento de Gastroenterología y Nutrición, Hospital Infantil de México Federico Gómez, Ciudad de México, México.

