



Chronic idiopathic constipation (CIC) negatively impacts quality of life and increases healthcare costs. Lubiprostone stimulates the secretion of intestinal fluid, in turn facilitating the passage of stools and alleviating associated symptoms. Lubiprostone has been available in Mexico since 2018, but its clinical efficacy has not been studied in a Mexican population.
AimTo evaluate the efficacy of lubiprostone, assessed by changes in spontaneous bowel movement (SBM) frequency after one week of treatment with 24 μg oral lubiprostone (b.i.d.), as well as its safety, over four weeks of treatment.
StudyRandomized, double-blind, placebo-controlled study on 211 adults with CIC in Mexico.
ResultsThe increase in SBM frequency, after one week of treatment, was significantly higher in the lubiprostone group than in the placebo group (mean: 4.9 [SD: 4.45] vs. 3.0 [3.14], p = 0.020). Secondary efficacy endpoints revealed a significantly higher proportion of SBM frequency/week in the lubiprostone group at weeks 2, 3, and 4. There was a better response within 24 h after the first dose with lubiprostone vs. placebo (60.0% vs. 41.5%; OR: 2.08, CI95%: [1.19, 3.62], p = 0.009) and the lubiprostone group also had significant improvement, with respect to straining, stool consistency, abdominal bloating, and Satisfaction Index. The main adverse events were gastrointestinal disorders in 13 (12.4%) lubiprostone-treated subjects and 4 (3.8%) control subjects.
ConclusionsOur data confirm the efficacy and safety of lubiprostone for the treatment of CIC in a Mexican population. Lubiprostone treatment induces relief from the most bothersome symptoms associated with constipation.
El estreñimiento crónico idiopático (ECI) impacta de manera negativa la calidad de vida e incrementa los costos de los sistemas de salud. La lubiprostona estimula la secreción de fluido intestinal, lo cual facilita el paso de las heces y alivia síntomas asociados. La lubiprostona ha estado disponible en México desde 2018, pero su eficacia clínica no ha sido estudiada en una población mexicana.
ObjetivoEvaluar la eficacia de la lubiprostona, por medio de la observación de los cambios en la frecuencia de evacuaciones espontáneas completas (EEC), después de una semana de tratamiento con 24 μg de lubiprostona oral dos veces al día, al igual que su seguridad, después de cuatro semanas de tratamiento.
EstudioEstudio aleatorizado, doble ciego, controlado con placebo, en 211 adultos con ECI en México.
ResultadosEl incremento de frecuencia de EEC tras una semana de tratamiento, fue significativamente más alto en el grupo de lubiprostona que en el grupo placebo (media: 4.9 [DE: 4.45] vs. 3.0 [3.14], p = 0.020). Los criterios de valoración secundarios de eficacia revelaron una proporción significativamente más alta de frecuencia EEC/semana en el grupo lubiprostona a las semanas 2, 3 y 4. Hubo una mejor respuesta dentro de 24 horas de la primera dosis de lubiprostona vs. placebo (60.0% vs. 41.5%; OR: 2.08, CI95%: [1.19, 3.62], p = 0.009), y el grupo de lubiprostona también presentó mejoría significativa respecto al pujo, la consistencia de las heces, la inflamación abdominal y el Índice de Satisfacción. Los principales eventos adversos fueron trastornos gastrointestinales en 13 (12.4%) de los sujetos tratados con lubiprostona y 4 (3.8%) de los sujetos control.
ConclusionesNuestros datos confirman la eficacia y seguridad de la lubiprostona para el tratamiento del ECI en una población mexicana. El tratamiento con lubiprostona induce alivio de los síntomas más molestos asociados con la constipación.
Chronic idiopathic constipation (CIC) is defined by the infrequent or difficult passage of stool with associated symptoms, such as abdominal bloating and discomfort, straining at defecation, and hard or lumpy stools. To make the diagnosis of CIC, predisposing conditions that could explain the symptoms must be ruled out, such as a low-fiber diet, sedentary lifestyle, drugs (particularly opiate analgesics and anticholinergic antidepressants), and bowel, neuromuscular, or metabolic disorders, as well as poor abdominal pressure or muscular atony.1,2 In CIC, symptoms result in part from abnormal colonic motility that delays intestinal content transit and hinders rectal emptying. The prevalence of constipation is highly variable (from 2% to 30%) and depends on many factors, including geographic location, ethnicity, sex, socioeconomic status, and the diagnostic criteria used.3 In Mexico, 14.4% of the general adult population has been reported to suffer from chronic constipation.4 Its impact on the quality of life of patients is certain, as well as the added costs associated with the condition. Currently, many pharmacologic therapies are available for the treatment of CIC and can be considered, in addition to dietary and lifestyle modifications. Osmotic laxatives (polyethylene-glycol and lactulose), stimulant laxatives (bisacodyl and sodium picosulfate), serotonergic agents (tegaserod, prucalopride), and prosecretory agents (secretagogues, such as lubiprostone, linaclotide, and plecanatide) are among the drug therapies with established efficacy and safety profiles.5 A significant number of studies supporting the therapeutic usefulness of lubiprostone, in particular, have been conducted. Lubiprostone is a bicyclic fatty acid metabolite analogue of prostaglandin E1 that activates the chloride channel 2 (ClC-2), located in the apical membrane of human intestinal epithelial cells, stimulating fluid secretion into the abdominal lumen.6,7 ClC-2 channel activation causes an increase in chloride ions (Cl−) and the subsequent secretion of intestinal fluid into the lumen, without altering serum sodium or potassium concentrations. By increasing intestinal fluid secretion, lubiprostone facilitates the passage of stool and alleviates symptoms associated with CIC. Systemic absorption of lubiprostone is virtually null and its unique measurable metabolite, M3, is not pharmacologically active; therefore, the risk of systemic toxicity and potential interaction with other drugs is minimal.
Additionally, ClC-2 channels play an important role in the restoration of tight junction complexes and the recovery of barrier function within the body. Lubiprostone has been shown to stimulate the recovery of mucosal barrier function in ex vivo studies of the intestine and colon, through the restoration of tight junction complexes.6,8 Safety and efficacy profiles of lubiprostone have been described in numerous clinical studies, and the level of evidence supports its recommendation for CIC treatment in clinical guidelines.4,9–12 Lubiprostone has shown superior efficacy over placebo, increasing stool frequency in the first week (5.69 vs. 3.46; p = 0.0001), reducing straining and improving stool consistency over all weeks (p ≤ 0.0003),13 as well as increasing the number of spontaneous bowel movements (SBMs) in the first week (5.89 vs. 3.99; p = 0.001).14 Lubiprostone was approved for use in patients with CIC by the Food and Drug Administration (FDA) in 2006 and by the European Medicines Agency (EMA) in 2012. However, clinical evidence from controlled studies using drugs to manage constipation in the Mexican population is limited.
We conducted a phase 3, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of 24 μg lubiprostone twice daily (BID) in Mexican subjects with CIC. While this clinical trial was performed to support the registration of lubiprostone in Mexico, our results provide additional evidence on the efficacy and safety of lubiprostone in the Mexican population, which was underrepresented in previous studies.
Material and methodsStudy design and participantsA phase 3, randomized, double-blind, parallel-group, placebo-controlled study was conducted at 10 sites across Mexico in Mexican adults (age >18 years) with functional constipation, according to the Rome III criteria, and a history of constipation defined as having SBMs with a frequency of <3 times per week on average for ≥6 months.15 Exclusion criteria were subjects that had constipation due to secondary causes (medications, hypothyroidism, depression) or those that had confirmed or suspected organic disorders of the large intestine or gastrointestinal surgery within three months prior to screening.
The study consisted of a 14-day screening period to confirm constipation symptoms, and a subsequent 4-week (28-day) double-blind treatment period, in which eligible subjects were randomized in a 1:1 ratio to receive either 24 µg of oral lubiprostone BID or placebo. The study was conducted between May 2016 and April 2017.
Study interventionsOnce informed consent was obtained, and before randomization, all participants completed a two-week screening period free of laxatives, to record their defecation behavior in a diary, confirming constipation. During that time, rescue medication (a bisacodyl tablet 5 mg or equivalent drug, or a glycerin suppository or equivalent drug, for those that did not respond to bisacodyl or its equivalent) was used if a subject had no adequate bowel movements for three consecutive days. Subjects whose constipation was confirmed during the screening period were randomized into a four-week double-blind treatment period to receive either lubiprostone or a matching placebo taken in one capsule orally BID with meals. Participants were asked to suspend use of all laxatives and prohibited drugs (including dietary fiber supplements), and to not change their diet or lifestyle habits, including their usual caffeine and alcohol intake, throughout the study. After starting the treatment period, participants returned to the study site on days 8, 15, and 29 for SBM frequency evaluation, as well as straining, stool consistency, abdominal symptom, and quality of life assessment. They were then followed for an additional 2 weeks, to determine safety outcomes when no treatment was administered (Fig. 1).

Forbidden concomitant medications included drugs with anticholinergic effects (except ipratropium bromide or any of its inhaled or nasal-spray forms), opioids, antispasmodics, cholinesterase inhibitors, antidiarrheal medications, anti-constipation medications (e.g., linaclotide), gastrointestinal prokinetic agents, laxative agents (e.g., PEG 3350), as well as homeopathic remedies, tricyclic antidepressants, or any medications known to relieve or cause constipation or constipation symptoms.
Endpoints and analysesThe primary endpoint was an increase in SBM frequency at week 1. Secondary endpoints included SBM frequency at weeks 2–4; the number of subjects that had a SBM within 24 h after the first dose; and mean degree of straining, stool consistency (Bristol Stool Form Scale), abdominal symptoms (bloating and discomfort assessed with a 5-point scale [a higher score indicating more severe symptoms]), and quality of life (established through a Patient Assessment of Constipation-Quality of Life Questionnaire [PAC-QOL] at weeks 1–4).
Safety endpointsThe safety endpoints evaluated the frequency and intensity of adverse events, as well as their relation to treatment. The Safety Analysis Set (SAS) included information on all participants that took at least one dose of the study drug or placebo.
Statistical analysisSample size was calculated at 204 (102 subjects in each group), based on the following assumptions: equal allocation, 90% power, a two-sided alpha of 0.05, placebo and treatment SBM frequency means of 4 and 5.9, respectively, a common standard deviation of 4 for SBM frequency at week 1 (using the Wilcoxon-Mann-Whitney test), and an assumed drop-out rate of 3% by day 4. Estimates of placebo and treatment responses were based on a previous study conducted by Sucampo Pharmaceuticals in the US (Protocol No. RTU/0211SC0131).
Patient consent and study ethicsThe present study was carried out in compliance with national regulations according to the principles stated in the Declaration of Helsinki the International Conference on the Harmonization Tripartite Guideline for Good Clinical Practice. The study protocol and its documents were approved by an Institutional Research and Ethics Committee, and by the national regulatory authority (Comisión Federal para la Protección contra Riesgos Sanitarios, COFEPRIS), with registry number 153300410A0196/2016. The study was registered at clinicaltrials.gov (NCT02729909).
Prior to enrollment, all participants received comprehensive information about the study and their participation, and all participants signed a written statement of informed consent.
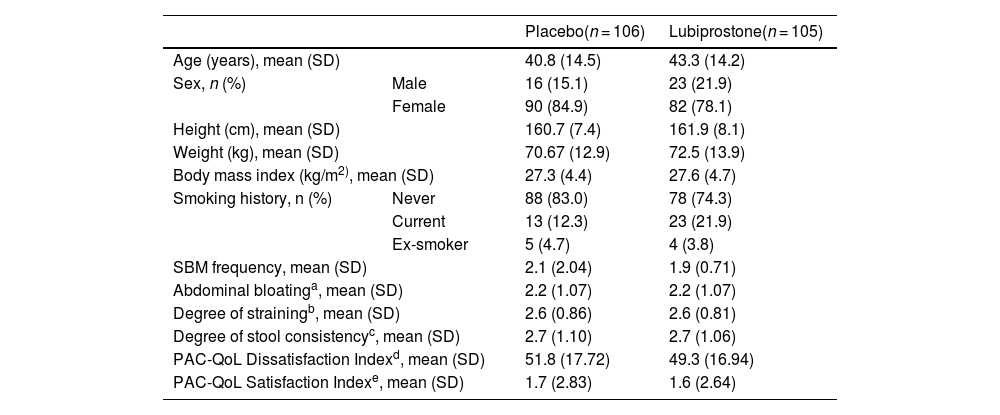
ResultsParticipantsA total of 211 subjects were enrolled in the study and received at least one dose of medication or placebo; 105 (49.8%) of the subjects were randomized into the lubiprostone group and 106 (50.2%) into the placebo group, representing the intention-to-treat (ITT) population, and their results are shown in this section. Six subjects discontinued the study (one in the lubiprostone group and five in the placebo group). Subject disposition is shown in Fig. 2, and the baseline demographic characteristics are presented in Table 1. No differences in the baseline characteristics were identified between groups.

Baseline demographic characteristics of subjects.
| Placebo(n = 106) | Lubiprostone(n = 105) | ||
|---|---|---|---|
| Age (years), mean (SD) | 40.8 (14.5) | 43.3 (14.2) | |
| Sex, n (%) | Male | 16 (15.1) | 23 (21.9) |
| Female | 90 (84.9) | 82 (78.1) | |
| Height (cm), mean (SD) | 160.7 (7.4) | 161.9 (8.1) | |
| Weight (kg), mean (SD) | 70.67 (12.9) | 72.5 (13.9) | |
| Body mass index (kg/m2), mean (SD) | 27.3 (4.4) | 27.6 (4.7) | |
| Smoking history, n (%) | Never | 88 (83.0) | 78 (74.3) |
| Current | 13 (12.3) | 23 (21.9) | |
| Ex-smoker | 5 (4.7) | 4 (3.8) | |
| SBM frequency, mean (SD) | 2.1 (2.04) | 1.9 (0.71) | |
| Abdominal bloatinga, mean (SD) | 2.2 (1.07) | 2.2 (1.07) | |
| Degree of strainingb, mean (SD) | 2.6 (0.86) | 2.6 (0.81) | |
| Degree of stool consistencyc, mean (SD) | 2.7 (1.10) | 2.7 (1.06) | |
| PAC-QoL Dissatisfaction Indexd, mean (SD) | 51.8 (17.72) | 49.3 (16.94) | |
| PAC-QoL Satisfaction Indexe, mean (SD) | 1.7 (2.83) | 1.6 (2.64) | |
SBM: spontaneous bowel movement; SD: Standard deviation.
Abdominal bloating is described using a 5-point scale ranging from 0 (None: No abdominal distension) to 4 (Very severe: Extremely strong abdominal distension).
The degree of straining is described using a 5-point scale ranging from 0 (no straining) to 4 (very strong straining).
The degree of stool consistency is described using a 7-point scale where decreasing values indicate harder stools.
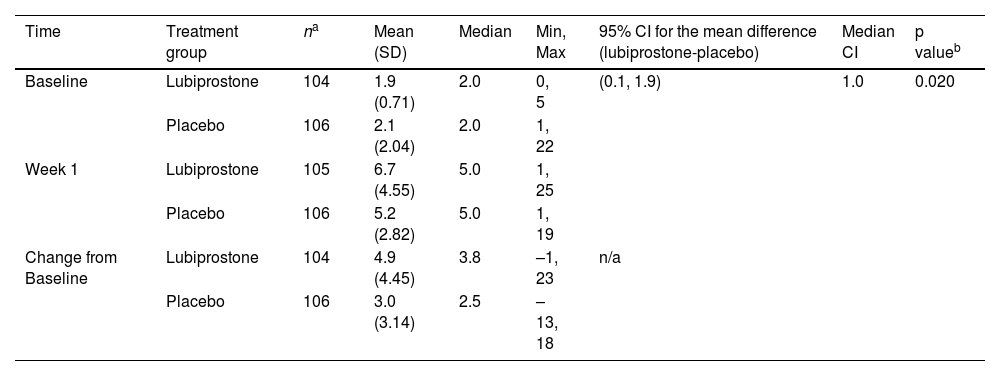
At treatment week 1, the mean (standard deviation [SD]) increase in SBM frequency from the baseline was significantly higher in the lubiprostone group (4.9 [4.45]) than in the placebo group (3.0 [3.14]) (p = 0.020) (Table 2).
Spontaneous bowel movement (SBM) frequency at week 1 (van Elteren test).
| Time | Treatment group | na | Mean (SD) | Median | Min, Max | 95% CI for the mean difference (lubiprostone-placebo) | Median CI | p valueb |
|---|---|---|---|---|---|---|---|---|
| Baseline | Lubiprostone | 104 | 1.9 (0.71) | 2.0 | 0, 5 | (0.1, 1.9) | 1.0 | 0.020 |
| Placebo | 106 | 2.1 (2.04) | 2.0 | 1, 22 | ||||
| Week 1 | Lubiprostone | 105 | 6.7 (4.55) | 5.0 | 1, 25 | |||
| Placebo | 106 | 5.2 (2.82) | 5.0 | 1, 19 | ||||
| Change from Baseline | Lubiprostone | 104 | 4.9 (4.45) | 3.8 | –1, 23 | n/a | ||
| Placebo | 106 | 3.0 (3.14) | 2.5 | –13, 18 | ||||
SD: standard deviation; 95% CI: 95% confidence interval; n/a: not applicable.
The secondary analysis of efficacy revealed a higher number of SBMs/week in the lubiprostone group at weeks 2, 3, and 4. All differences were statistically significant, when compared with the placebo group (mean [SD] in lubiprostone vs. placebo at week 2: 6.9 [4.40] vs. 5.5 [2.80], p = 0.043; week 3: 7.6 [4.53] vs. 5.9 [3.87], p = 0.003; and week 4: 7.1 [4.30] vs. 5.6 [3.52], p = 0.013).
The number of subjects that responded with a SBM within 24 h after the first dose was also higher with lubiprostone treatment versus placebo (63/105 [60.0%] vs. 44/106 [41.5%]; OR 2.08, [95% CI 1.19, 3.62], p = 0.009).
Significant reductions in the mean degree of straining were also noted with lubiprostone treatment at weeks 1, 2, 3, and 4 (mean [SD] in lubiprostone vs. placebo at week 1: 4.1 [1.3] vs. 3.4 [1.08], p < 0.001; week 2: 4.1 [1.13] vs. 3.5 [1.05], p < 0.001; week 3: 4.1 [1.12] vs. 3.5 [0.93], p < 0.001; and week 4: 4.1 [1.19] vs. 3.6 [0.93], p < 0.001) (Fig. 3A).
 and a bdominal bloating (B) during the study.")
Statistically significant improvements (change from baseline) in stool consistency, as assessed by the Bristol Stool Form Scale, showing a weekly positive change in the scale, were noted with lubiprostone treatment at weeks 1, 2, 3, and 4 (mean [SD] in lubiprostone vs. placebo at week 1: +2.0 [0.88] vs. +1.6 [0.85], p = 0.001; week 2: +1.8 [0.93] vs. +1.5 [0.89], p = 0.004; week 3: +1.6 [0.92] vs. +1.3 [0.82], p = 0.024; and week 4: +1.5 [0.86] vs. +1.2 [0.92], p = 0.010) (Fig. 3B).
A significant reduction in abdominal bloating and discomfort was observed with lubiprostone treatment. Those improvements were statistically significant at weeks 3 and 4 (mean abdominal bloating [SD] in lubiprostone vs. placebo at week 3: 1.0 [1.01] vs. 1.4 [0.93], p < 0.001; week 4: 0.9 [0.94] vs. 1.2 [0.98], p = 0.004).
The Satisfaction Index (evaluated through abdominal discomfort) was higher on weeks 2 and 4 for the lubiprostone treatment group, when compared with the placebo treatment group (mean [SD] in lubiprostone vs. placebo at week 2: 7.3 [3.78] vs. 5.2 [3.68], p < 0.001; week 4: 9.0 [3.98] vs. 6.5 [4.43], p < 0.001).
An exploratory analysis to determine the number of SBMs per week with the sensation of complete evacuation was conducted. The results showed a higher number of SBMs/week with the sensation of complete evacuation at weeks 1, 2, 3, and 4 for lubiprostone over placebo, but without statistical significance (data not shown).
Rescue medicationA total of 38 participants required rescue medication (more than 72 h with no SBMs); 19 (50.0%) in the placebo group and 19 (50.0%) in the lubiprostone group (p > 0.05). The two main medications used for rescue in both groups were glycerin suppository (placebo: 13 [59.1%]; lubiprostone: 17 [70.8%], p > 0.05) and bisacodyl 5 mg (placebo: 2 [9.1%]; lubiprostone: 2 [8.3%], p > 0.05).
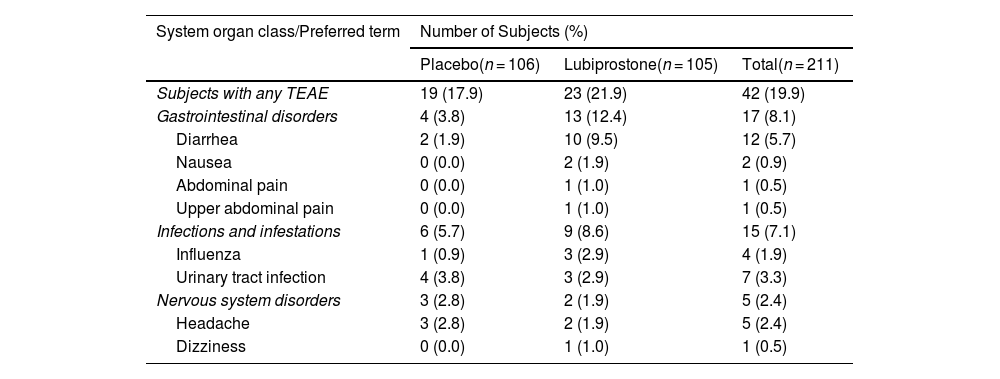
Safety endpointsA total of 63 Treatment-Emergent Adverse Events (TEAEs) were reported by 42 subjects (19.9%); 23 subjects (21.9%) in the lubiprostone group and 19 (17.9%) in the placebo group (Table 3). There were no deaths and no serious adverse events reported during the study. The largest numbers of adverse events were reported by 17 subjects (8.1%) in System Organ Class (SOC) Gastrointestinal disorders (reported by 13 [12.4%] and 4 [3.8%] subjects in the lubiprostone and placebo treatment groups, respectively), and SOC Infections and infestations (15 subjects (7.1%), 9 (8.6%) and 6 (5.7%) subjects in the lubiprostone and placebo groups, respectively). There were no adverse events that led to treatment interruptions or withdrawal from the study.
Treatment-emergent adverse events (TEAE) by system organ class (SOC) and preferred term (PT).
| System organ class/Preferred term | Number of Subjects (%) | ||
|---|---|---|---|
| Placebo(n = 106) | Lubiprostone(n = 105) | Total(n = 211) | |
| Subjects with any TEAE | 19 (17.9) | 23 (21.9) | 42 (19.9) |
| Gastrointestinal disorders | 4 (3.8) | 13 (12.4) | 17 (8.1) |
| Diarrhea | 2 (1.9) | 10 (9.5) | 12 (5.7) |
| Nausea | 0 (0.0) | 2 (1.9) | 2 (0.9) |
| Abdominal pain | 0 (0.0) | 1 (1.0) | 1 (0.5) |
| Upper abdominal pain | 0 (0.0) | 1 (1.0) | 1 (0.5) |
| Infections and infestations | 6 (5.7) | 9 (8.6) | 15 (7.1) |
| Influenza | 1 (0.9) | 3 (2.9) | 4 (1.9) |
| Urinary tract infection | 4 (3.8) | 3 (2.9) | 7 (3.3) |
| Nervous system disorders | 3 (2.8) | 2 (1.9) | 5 (2.4) |
| Headache | 3 (2.8) | 2 (1.9) | 5 (2.4) |
| Dizziness | 0 (0.0) | 1 (1.0) | 1 (0.5) |
A treatment-emergent adverse event (TEAE) is defined as an AE whose date of onset occurs after the first dose of study drug through the follow-up visit.
Subjects with one or more adverse events within a level of the Medical Dictionary for Regulatory Activities (MedDRA) term are counted only once in that level. Percentages are based on the number of subjects in the safety set for each treatment group.
SOC terms are sorted in alphabetical order and PT are sorted in decreasing frequency in the lubiprostone group.
MedDRA Dictionary (Version 20.0 Mixed) was used for coding adverse events.
Overall, all adverse events were mild and transient. Diarrhea was more frequently reported in the lubiprostone treatment group than in the placebo group (10 subjects [9.5%] vs. 2 subjects [1.9%], respectively) and was often described as related to study drug administration. Reported infections and infestations included urinary tract infections (3 subjects [2.9%] in the lubiprostone treatment group and 4 subjects [3.8%] in the placebo treatment group) and influenza (3 subjects [2.9%] in the lubiprostone treatment group and 1 subject [0.9%] in the placebo treatment group); all the TEAEs were considered unrelated to study drug administration. Common adverse events reported as study drug-related were consistent with the known safety profile of lubiprostone, and no new safety signals were identified in the present study.
DiscussionLubiprostone has been reported in previous studies to be safe and effective for the treatment of CIC. Our study conducted on Mexican CIC subjects showed that 24 μg of lubiprostone BID was more effective than placebo, as measured by SBM frequency at week one of treatment, the primary endpoint. That statistically significant improvement was also maintained over a 4-week period. Johanson et al.13 reported a significant improvement in constipation-related symptoms that was replicated by Barish et al.14 We corroborated herein sustained improvement from the baseline, in symptoms including straining, stool consistency, and abdominal bloating and discomfort. Those improvements were statistically significant at weeks 1, 2, 3, and 4 for straining and stool consistency, at weeks 1, 3, and 4 for abdominal bloating, and at weeks 3 and 4 for abdominal discomfort.
Regarding improvement of symptoms associated with constipation (abdominal pain, discomfort), which were significantly reduced in the group treated with lubiprostone, they could be related to changes in the local intestinal inflammatory mediators that are modified when intestinal transit increases. We saw a higher increase in the Bristol Stool Form Scale in patients receiving lubiprostone, compared with placebo, that was observed throughout the study, revealing stool softening as another benefit of lubiprostone use.
Ours is the first study evaluating lubiprostone efficacy and safety in a Latin American population, and interestingly, the incidence of adverse events was lower than that reported in previous studies conducted on other populations.13,14
The fact that changes in local intraluminal conditions can affect the intestinal microbiome, with subsequent changes in inflammatory mediators that could impact the subjective parameters evaluated, cannot be ruled out. Additional studies are required to determine the role of lubiprostone in shaping the intestinal microbiome and its clinical implications.
Lubiprostone was well tolerated and the commonly reported adverse events were largely consistent with its known safety profile. Interestingly, the incidence of nausea in the lubiprostone group was lower, compared with that reported in previous studies. Whether that is relevant or not should be determined through careful and continuous patient assessment during treatment with the drug.
Although it was not a primary endpoint of our study, an exploratory analysis on the number of SBMs per week with the sensation of complete evacuation revealed sustained improvement with lubiprostone throughout the study, providing insight for future clinical studies that could be conducted using said parameter as one of the primary endpoints.
Even though the present study was carried out using the Rome III diagnostic criteria for constipation, changes implemented in the Rome IV criteria would modify practically none of the parameters of admission or evaluation of this work, so our results can be considered valid and useful under the current definitions (Rome IV), but they could be re-evaluated in detail in case of future changes in the diagnostic and therapeutic criteria for CIC.
One of the limitations of our study was its duration, but a similar previous study of up to 48 weeks showed that the greatest changes occurred within the first six weeks and that outcomes were not modified much later.16 In addition, the prevalence of complete SBMs (those that provide complete emptying satisfaction, in the absence of laxative use) was not evaluated as an outcome, because it has recently been proposed as an efficacy criterion. Thus, it should be considered for future studies on the efficacy of CIC treatments.
Regarding safety, the fact that no participant dropped out of the present study for reasons related to safety was relevant. Moreover, when compared with previous reports,11,13,14 a lower number of participants experienced nausea during our study.
In conclusion, our data are consistent with those of previous reports, showing that lubiprostone, at a dose level of 24 µg BID, was a safe and effective treatment for constipation in a Mexican population, confirming its usefulness in the treatment of CIC.
Financial disclosureThe present study has been funded by Takeda Development Center Americas INC. Medical writing support was provided by iLS Clinical Research SC and funded by Takeda Mexico.
Conflict of interestE. Coss-Adame has served on the Advisory Board of Takeda and Asofarma. He has been a consultant for Alfasigma, Takeda Mexico, Asofarma Mexico. He has also been a speaker for Alfasigma, Takeda Mexico, Carnot, Sanfer, and Endomedica.
J.M. Remes Troche has served on the Advisory Board of Takeda and Asofarma. He has been a consultant for Alfasigma, Takeda Mexico, Asofarma Mexico, Bicodex and Sanfer. He has also been a speaker for Alfasigma, Takeda Mexico, Carnot, Sanfer, and Endomedica.
R. Flores Rendón, has no conflict of interest to declare.
J. L. Tamayo de la Cuesta, has no conflict of interest to declare.
Miguel Ángel Valdovinos Díaz, has served on the Advisory Board of Takeda and Biocodex. He has been a speaker for Takeda, Carnot, Mayoly, and Biocodex.
The funding, design, conduction, supervision, and report of this study was done by the sponsors, Takeda Development Center America Inc. and Takeda Mexico, S.A. de C.V. Medical writing support, under the guidance of the authors, was provided by Mauricio Rodríguez-Álvarez, MD and iLS Clinical Research, S.C., and was funded by Takeda Mexico, S.A. de C.V. in accordance with Good Publication Practices (GPP3) guidelines.
Medical writing support, technical comments, and manuscript formatting, under the supervision of the authors, was provided by Carlos A. Díaz-Tufinio and iLS Clinical Research, S.C.

